Oční kapky: Lumigan 3ml Použití, vedlejší účinky a dávkování. Cena v internetové lékárně. Generické léky bez předpisu.
Co je Lumigan a jak se používá?
Lumigan 3ml je lék na předpis používaný k léčbě příznaků zvýšeného nitroočního tlaku způsobeného určitými typy glaukomu a hypotrichózy řas. Lumigan lze užívat samostatně nebo s jinými léky.
Lumigan patří do třídy léků nazývaných Antiglaukoma, agonisté prostaglandinu.
Není známo, zda je přípravek Lumigan bezpečný a účinný u dětí.
Jaké jsou možné vedlejší účinky přípravku Lumigan?
Lumigan může způsobit závažné nežádoucí účinky včetně:
- otok očí,
- zarudnutí oka,
- silné nepohodlí v oku,
- krusta nebo drenáž z oka,
- změny vidění a
- červená, oteklá nebo svědící víčka
Pokud máte některý z výše uvedených příznaků, okamžitě vyhledejte lékařskou pomoc.
Mezi nejčastější vedlejší účinky Lumiganu patří:
- zarudnutí očí
Informujte lékaře, pokud máte jakýkoli nežádoucí účinek, který vás obtěžuje nebo který neustupuje.
To nejsou všechny možné vedlejší účinky přípravku Lumigan. Pro více informací se zeptejte svého lékaře nebo lékárníka.
Zavolejte svého lékaře o radu ohledně nežádoucích účinků. Nežádoucí účinky můžete hlásit úřadu FDA na čísle 1-800-FDA-1088.
POPIS
LUMIGAN® (bimatoprost oční roztok) 0,03% je syntetický analog prostamidu s oční hypotenzní aktivitou. Jeho chemický název je (Z)-7-[(1R,2R,3R,5S)-3,5-dihydroxy-2[(1E,3S)-3-hydroxy-5-fenyl-1-pentenyl]cyklopentyl]- 5-N-ethylheptenamid a jeho molekulová hmotnost je 415,58. Jeho molekulový vzorec je C25H37NO4. Jeho chemická struktura je:
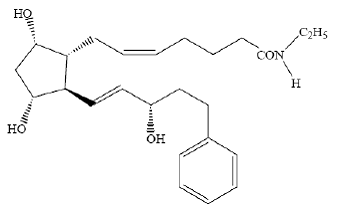
Bimatoprost je prášek, který je velmi dobře rozpustný v ethylalkoholu a methylalkoholu a mírně rozpustný ve vodě. LUMIGAN® 0,03 % je čirý, izotonický, bezbarvý, sterilní oční roztok s osmolalitou přibližně 290 mOsmol/kg.
LUMIGAN® 0,03 % obsahuje Aktivní: bimatoprost 0,3 mg/ml; Neaktivní: benzalkoniumchlorid 0,05 mg/ml; chlorid sodný; hydrogenfosforečnan sodný; kyselina citronová; a vyčištěná voda. Pro úpravu pH lze přidat hydroxid sodný a/nebo kyselinu chlorovodíkovou. Hodnota pH během doby trvanlivosti se pohybuje v rozmezí 6,8-7,8.
INDIKACE
LUMIGAN® (bimatoprost oční roztok) 0,01% je indikován ke snížení zvýšeného nitroočního tlaku u pacientů s glaukomem s otevřeným úhlem nebo oční hypertenzí.
DÁVKOVÁNÍ A PODÁVÁNÍ
Doporučená dávka je jedna kapka do postiženého oka (očí) jednou denně večer. LUMIGAN® (bimatoprost oční roztok) 0,01% by neměl být podáván více než jednou denně, protože se ukázalo, že častější podávání analogů prostaglandinu může snížit účinek snížení nitroočního tlaku.
Snížení nitroočního tlaku začíná přibližně 4 hodiny po prvním podání a maximálního účinku je dosaženo přibližně za 8 až 12 hodin.
LUMIGAN® 0,01 % lze používat současně s jinými topickými oftalmickými léčivými přípravky ke snížení nitroočního tlaku. Pokud se používá více než jeden topický oční lék, měly by být léky podávány s odstupem alespoň pěti (5) minut.
JAK DODÁVÁNO
Dávkové formy A Síly
Oční roztok obsahující bimatoprost 0,1 mg/ml.
Skladování a předávání
LUMIGAN® (bimatoprost oční roztok) 0,01% se dodává sterilní v neprůhledných bílých polyethylenových očních lahvičkách a špičkách s tyrkysovým polystyrénovým uzávěrem v následujících velikostech:
2,5 ml naplňte do 5 ml nádoby - NDC 0023-3205-03 5 ml náplň do 10 ml nádoby - NDC 0023-3205-05 7,5 ml náplň do 10 ml nádoby - NDC 0023-3205-08
Úložný prostor: Uchovávejte při teplotě 2 °C až 25 °C (36 °F až 77 °F).
Po otevření lze LUMIGAN® 0,01 % používat do data expirace uvedeného na lahvičce.
Distribuce: Allergan USA, Inc. Madison, NJ 07940. Revize: březen 2022
VEDLEJŠÍ EFEKTY
Následující nežádoucí účinky jsou popsány jinde v označení:
- Pigmentace včetně blefarální pigmentace a hyperpigmentace duhovky [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]
- Změny řas [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]
- Nitrooční záněty [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]
- Makulární edém [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]
- Přecitlivělost [viz KONTRAINDIKACE ]
Zkušenosti z klinických studií
Vzhledem k tomu, že klinické studie jsou prováděny za velmi odlišných podmínek, nelze míry nežádoucích reakcí pozorované v klinických studiích léku přímo srovnávat s mírami v klinických studiích jiného léku a nemusí odrážet míry pozorované v praxi.
Ve 12měsíční klinické studii s očními roztoky bimatoprostu 0,01 % byla nejčastějším nežádoucím účinkem spojivková hyperémie (31 %). Přibližně 1,6 % pacientů přerušilo léčbu kvůli spojivkové hyperémii. Jiné nežádoucí účinky (hlášené u 1 až 4 % pacientů) s LUMIGAN® 0,01 % v této studii zahrnovalo otok spojivek, spojivkové krvácení, podráždění oka, bolest oka, svědění oka, erytém očního víčka, svědění očních víček, růst řas, hypertrichózu, podráždění v místě aplikace, tečkovitou keratitidu, hyperpigmentaci kůže, rozmazané vidění a ostrost vidění snížena.
Postmarketingové zkušenosti
Následující nežádoucí účinky byly zjištěny během užívání po schválení LUMIGAN® 0,01 %. Protože jsou tyto reakce hlášeny dobrovolně z populace nejisté velikosti, není vždy možné spolehlivě odhadnout jejich frekvenci nebo stanovit příčinnou souvislost s expozicí léku. Tyto reakce, které byly vybrány k zahrnutí buď kvůli jejich závažnosti, četnosti hlášení, možné kauzální souvislosti s LUMIGAN® nebo kombinace těchto faktorů zahrnují: příznaky podobné astmatu, závratě, suché oko, dušnost, výtok z oka, otok oka, pocit cizího tělesa, bolest hlavy, přecitlivělost včetně známek a příznaků oční alergie a alergické dermatitidy, hypertenze, zvýšené slzení, periorbitální změny a změny víček včetně prohloubení rýhy očního víčka a fotofobie.
DROGOVÉ INTERAKCE
Nebyly poskytnuty žádné informace
VAROVÁNÍ
Zahrnuto jako součást "OPATŘENÍ" Sekce
OPATŘENÍ
Pigmentace
Bylo hlášeno, že oční roztok bimatoprostu způsobuje změny pigmentovaných tkání. Nejčastěji hlášenými změnami byla zvýšená pigmentace duhovky, periorbitální tkáně (očního víčka) a řas. Očekává se, že pigmentace se bude zvyšovat, dokud je bimatoprost podáván. Změna pigmentace je způsobena spíše zvýšeným obsahem melaninu v melanocytech než zvýšením počtu melanocytů. Po vysazení bimatoprostu bude pigmentace duhovky pravděpodobně trvalá, zatímco pigmentace periorbitální tkáně a změny řas byly u některých pacientů reverzibilní. Pacienti, kteří jsou léčeni, by měli být informováni o možnosti zvýšené pigmentace. Dlouhodobé účinky zvýšené pigmentace nejsou známy.
Změna barvy duhovky nemusí být patrná po několik měsíců až let. Typicky se hnědá pigmentace kolem zornice šíří soustředně směrem k periferii duhovky a celá duhovka nebo její části se zhnědnou. Zdá se, že léčba neovlivňuje névy ani pihy duhovky. Zatímco léčba LUMIGAN® (oční roztok bimatoprostu) 0,01% může pokračovat u pacientů, u kterých se rozvine znatelně zvýšená pigmentace duhovky, tito pacienti by měli být pravidelně vyšetřováni.
Změny řas
LUMIGAN® 0,01 % může postupně měnit řasy a vellus v ošetřeném oku. Tyto změny zahrnují zvýšenou délku, tloušťku a počet řas. Změny řas jsou po přerušení léčby obvykle reverzibilní.
Nitrooční zánět
Bylo hlášeno, že analogy prostaglandinu, včetně bimatoprostu, způsobují nitrooční zánět. Navíc, protože tyto přípravky mohou zhoršit zánět, je třeba opatrnosti u pacientů s aktivním nitroočním zánětem (např. uveitida).
Makulární edém
Během léčby očním roztokem bimatoprost byl hlášen makulární edém, včetně cystoidního makulárního edému. LUMIGAN® 0,01 % by měl být používán s opatrností u afakických pacientů, u pseudofakických pacientů s natrženým zadním pouzdrem čočky nebo u pacientů se známými rizikovými faktory pro makulární edém.
Bakteriální keratitida
Byly hlášeny případy bakteriální keratitidy spojené s používáním vícedávkových obalů topických oftalmologických přípravků. Tyto nádoby byly neúmyslně kontaminovány pacienty, kteří ve většině případů měli souběžné onemocnění rohovky nebo narušení očního epiteliálního povrchu.
Použití kontaktních čoček
LUMIGAN® 0,01% obsahuje benzalkoniumchlorid, který může být absorbován měkkými kontaktními čočkami a může způsobit změnu barvy. Kontaktní čočky je třeba před nakapáním LUMIGAN® 0,01 % vyjmout a lze je znovu nasadit 15 minut po jeho podání.
Neklinická toxikologie
Karcinogeneze, mutageneze, zhoršení plodnosti
Karcinogeneze
Bimatoprost nebyl karcinogenní ani u myší, ani u potkanů, když byl podáván perorální sondou po dobu 104 týdnů v dávkách až 2 mg/kg/den a 1 mg/kg/den (192 a 291násobek odhadované systémové expozice u člověka bimatoprostu 0,03 % dávkování oboustranně jednou denně, v daném pořadí, na základě hladin AUC v krvi)
Mutageneze
Bimatoprost nebyl mutagenní ani klastogenní v Amesově testu, v testu myšího lymfomu ani v in vivo mikronukleárních testech u myší.
Zhoršení Plodnosti
Bimatoprost nezhoršil fertilitu u samců nebo samic potkanů až do dávek 0,6 mg/kg/den (nejméně 103násobek doporučené lidské expozice bimatoprostu 0,03 % podávanému bilaterálně jednou denně na základě hladin AUC v krvi).
Použití u konkrétních populací
Těhotenství
Shrnutí rizik
Neexistují žádné adekvátní a dobře kontrolované studie podávání LUMIGAN® (bimatoprost oční roztok) 0,01% u těhotných žen. Na základě postmarketingových zkušeností s bimatoprostem nedochází k žádnému zvýšení rizika závažných vrozených vad nebo potratů.
V embryofetálních vývojových studiích vedlo podávání bimatoprostu březím myším a potkanům během organogeneze k potratu a předčasnému porodu při perorálních dávkách nejméně 33násobných (myši) nebo 94násobných (krysy), než je expozice člověka bimatoprostu 0,03 % podávanému bilaterálně jednou denně ( na základě hladiny krevní oblasti pod křivkou [AUC]). Tyto nežádoucí účinky nebyly pozorovány při 2,6násobku (myši) a 47násobku (krysy) expozice člověka bimatoprostu 0,03% podávanému bilaterálně jednou denně (na základě hladin AUC v krvi).
Ve studiích pre/postnatálního vývoje vedlo podávání bimatoprostu březím krysám od organogeneze do konce laktace ke zkrácení délky březosti a tělesné hmotnosti plodu a zvýšené úmrtnosti plodů a mláďat při perorálních dávkách alespoň 41násobku systémové expozice bimatoprostu u člověka 0,03 % dávkované bilaterálně jednou denně (na základě hladin AUC v krvi). Nebyly pozorovány žádné nežádoucí účinky u potomků potkanů při expozicích odhadovaných na 14násobek expozice člověka bimatoprostu 0,03% podávanému bilaterálně jednou denně (na základě hladin AUC v krvi).
Vzhledem k tomu, že reprodukční studie na zvířatech ne vždy předpovídají lidskou odpověď, měl by být LUMIGAN® 0,01 % podáván během těhotenství pouze v případě, že potenciální přínos ospravedlňuje potenciální riziko pro plod.
Data
Údaje o zvířatech
Ve studii embryofetálního vývoje na potkanech byl u březích potkaních samic pozorován potrat, kterému byl během organogeneze perorálně podáván bimatoprost v dávce 0,6 mg/kg/den (94násobek systémové expozice člověka bimatoprostu 0,03 % podávanému bilaterálně jednou denně, na základě AUC). Hladina bez pozorovaných nepříznivých účinků (NOAEL) pro potrat byla 0,3 mg/kg/den (odhadovaná na 47násobek systémové expozice člověka bimatoprostu 0,03 % podávanému bilaterálně jednou denně na základě AUC). U potkaních plodů nebyly pozorovány žádné abnormality při dávkách do 0,6 mg/kg/den.
Ve studii embryofetálního vývoje na myších byly u březích myší pozorovány potraty a předčasný porod u březích myší, kterým byl během organogeneze perorálně podáván bimatoprost v dávkách vyšších nebo rovných 0,3 mg/kg/den (33násobek systémové expozice člověka bimatoprostu 0,03 % podávanému bilaterálně jednou denně, na základě AUC). NOAEL pro potrat a předčasný porod byla 0,1 mg/kg/den (2,6násobek systémové expozice u člověka 0,03% bimatoprostu podávanému bilaterálně jednou denně, na základě AUC). U myších plodů nebyly pozorovány žádné abnormality při dávkách až 0,6 mg/kg/den (72násobek systémové expozice u člověka 0,03% bimatoprostu podávanému bilaterálně jednou denně, na základě AUC).
Ve studii prenatálního/postnatálního vývoje vedla léčba březích potkanů bimatoprostem perorálně od 7. dne březosti do 20. dne laktace ke zkrácení délky březosti, zvýšené pozdní resorpci, úmrtí plodu a postnatální úmrtnosti mláďat a snížení tělesné hmotnosti mláďat při dávkách vyšších než nebo rovné 0,3 mg/kg/den. Tyto účinky byly pozorovány při expozicích nejméně 41násobku systémové expozice u člověka bimatoprostu 0,03% podávanému bilaterálně jednou denně, na základě AUC. NOAEL pro postnatální vývoj a páření potomstva byla 0,1 mg/kg/den (odhadovaná na 14násobek systémové expozice člověka bimatoprostu 0,03% podávanému bilaterálně jednou denně, na základě AUC).
Laktace
Shrnutí rizik
Není známo, zda by lokální oční léčba LUMIGAN® 0,01 % mohla vést k dostatečné systémové absorpci k produkci detekovatelných množství v mateřském mléce. Ve studiích na zvířatech bylo prokázáno, že bimatoprost je přítomen v mateřském mléce kojících potkanů při intravenózní dávce (tj. 1 mg/kg) 970násobku RHOD (na základě mg/rn 2), avšak nejsou k dispozici žádné údaje na zvířatech v klinicky relevantních dávkách.
Vývojové a zdravotní přínosy kojení by měly být zváženy spolu s klinickou potřebou matky LUMIGAN® 0,01 % a případnými nežádoucími účinky na kojené dítě LUMIGAN® 0,01 %.
Pediatrické použití
Použití u pediatrických pacientů mladších 16 let se nedoporučuje z důvodu potenciálních bezpečnostních obav souvisejících se zvýšenou pigmentací po dlouhodobém chronickém užívání.
Geriatrické použití
Mezi staršími a jinými dospělými pacienty nebyly pozorovány žádné celkové klinické rozdíly v bezpečnosti nebo účinnosti.
PŘEDÁVKOVAT
Nejsou dostupné žádné informace o předávkování u lidí. Pokud dojde k předávkování LUMIGAN® (oční roztok bimatoprostu) 0,01 %, léčba by měla být symptomatická.
Ve studiích na myších a potkanech podávaných perorálně (žaludeční sondou) dávky až do 100 mg/kg/den nevyvolaly žádnou toxicitu. Tato dávka vyjádřená v mg/m2 je nejméně 210krát vyšší než náhodná dávka jedné lahvičky LUMIGAN® 0,01 % pro 10 kg dítě.
KONTRAINDIKACE
LUMIGAN® 0,01% je kontraindikován u pacientů s přecitlivělostí na bimatoprost nebo na kteroukoli složku přípravku (viz NEŽÁDOUCÍ REAKCE ].
KLINICKÁ FARMAKOLOGIE
Mechanismus působení
Bimatoprost, analog prostaglandinu, je syntetický strukturní analog prostaglandinu s oční hypotenzní aktivitou. Selektivně napodobuje účinky přirozeně se vyskytujících látek, prostamidů. Předpokládá se, že bimatoprost snižuje nitrooční tlak (IOP) u lidí zvýšením odtoku komorové vody jak trabekulární síťovinou, tak uveosklerální cestou. Zvýšený NOT představuje hlavní rizikový faktor ztráty pole glaukomu. Čím vyšší je hladina IOP, tím větší je pravděpodobnost poškození zrakového nervu a ztráty zorného pole.
Farmakokinetika
Vstřebávání
Poté, co byla jedna kapka očního roztoku bimatoprostu 0,03% podávána jednou denně do obou očí 15 zdravým subjektům po dobu dvou týdnů, koncentrace v krvi dosáhla vrcholu během 10 minut po podání dávky a byla pod spodním limitem detekce (0,025 ng/ml) u většiny subjektů v rámci 1,5 hodiny po podání. Průměrné hodnoty Cmax a AUC0-24h byly podobné 7. a 14. den přibližně 0,08 ng/ml a 0,09 ng•h/ml, což ukazuje, že ustáleného stavu bylo dosaženo během prvního týdne očního podávání. V průběhu času nedošlo k žádné významné systémové akumulaci léčiva.
Rozdělení
Bimatoprost je středně distribuován do tělesných tkání s distribučním objemem v ustáleném stavu 0,67 l/kg. V lidské krvi se bimatoprost nachází hlavně v plazmě. Přibližně 12 % bimatoprostu zůstává nevázáno v lidské plazmě.
Odstranění
Metabolismus
Bimatoprost je hlavní cirkulující látkou v krvi, jakmile se po očním podání dostane do systémové cirkulace. Bimatoprost poté podléhá oxidaci, N-deethylaci a glukuronidaci za vzniku různých metabolitů.
Vylučování
Po intravenózní dávce radioaktivně značeného bimatoprostu (3,12 mcg/kg) šesti zdravým subjektům byla maximální koncentrace nezměněného léku v krvi 12,2 ng/ml a rychle klesala s poločasem eliminace přibližně 45 minut. Celková clearance bimatoprostu z krve byla 1,5 l/hod/kg. Až 67 % podané dávky bylo vyloučeno močí, zatímco 25 % dávky bylo nalezeno ve stolici.
Klinické studie
Ve 12měsíční klinické studii pacientů s glaukomem s otevřeným úhlem nebo oční hypertenzí s průměrnou výchozí hodnotou NOT 23,5 mmHg byl účinek LUMIGAN® 0,01 % jednou denně (večer) na snížení NOT až 7,5 mmHg.
INFORMACE PRO PACIENTA
Potenciál pro pigmentaci
Informujte pacienty o možnosti zvýšené hnědé pigmentace duhovky, která může být trvalá. Také informujte pacienty o možnosti ztmavnutí kůže očních víček, které může být reverzibilní po vysazení LUMIGAN® (bimatoprost oční roztok) 0,01%.
Potenciál pro změny řas
Informujte pacienty o možnosti změn řas a vellusu na ošetřovaném oku během léčby LUMIGAN® 0,01%. Tyto změny mohou vést k rozdílům mezi očima v délce, tloušťce, pigmentaci, počtu řas nebo vellus chloupků a/nebo směru růstu řas. Změny řas jsou po přerušení léčby obvykle reverzibilní.
Manipulace s kontejnerem
Poučte pacienty, aby se špička dávkovacího zásobníku nedostala do kontaktu s okem, okolními strukturami, prsty nebo jakýmkoli jiným povrchem, aby se zabránilo kontaminaci roztoku běžnými bakteriemi, o nichž je známo, že způsobují oční infekce. Použitím kontaminovaných roztoků může dojít k vážnému poškození oka a následné ztrátě zraku.
Kdy vyhledat radu lékaře
Informujte pacienty, že pokud se u nich rozvine interkurentní oční stav (např. trauma nebo infekce), podstoupí oční operaci nebo se u nich rozvinou jakékoli oční reakce, zejména zánět spojivek a reakce očních víček, měli by okamžitě vyhledat radu svého lékaře ohledně dalšího používání LUMIGAN® 0,01% .
Použití kontaktních čoček
Informujte pacienty, že LUMIGAN® 0,01 % obsahuje benzalkoniumchlorid, který může být absorbován měkkými kontaktními čočkami. Kontaktní čočky je třeba před nakapáním LUMIGAN® 0,01 % vyjmout a lze je znovu nasadit 15 minut po jeho podání.
Použití s jinými očními léky
Informujte pacienty, že pokud se používá více než jeden topický oftalmologický lék, léky by měly být podávány alespoň pět (5) minut mezi aplikacemi.