Zetia 10mg Ezetimibe Použití, vedlejší účinky a dávkování. Cena v internetové lékárně. Generické léky bez předpisu.
Co je Zetia a jak se používá?
Zetia je lék na předpis používaný k léčbě příznaků vysokého cholesterolu a ke snížení cholesterolu. Zetia lze užívat samostatně nebo s jinými léky.
Zetia patří do třídy léků nazývaných látky snižující hladinu lipidů, 2-azetidinony.
Není známo, zda je přípravek Zetia 10 mg bezpečný a účinný u dětí mladších 10 let.
Jaké jsou možné vedlejší účinky přípravku Zetia?
Zetia může způsobit závažné nežádoucí účinky včetně:
- bolest svalů,
- citlivost nebo slabost svalů,
- horečka,
- neobvyklá únava a
- tmavě zbarvená moč
Pokud máte některý z výše uvedených příznaků, okamžitě vyhledejte lékařskou pomoc.
Mezi nejčastější nežádoucí účinky přípravku Zetia 10 mg patří:
- bolesti svalů nebo kloubů,
- ucpaný nos,
- bolest dutin,
- bolest krku,
- průjem a
- bolest v ruce nebo noze
Informujte svého lékaře, pokud máte jakýkoli nežádoucí účinek, který vás obtěžuje nebo který neustupuje.
To nejsou všechny možné vedlejší účinky přípravku Zetia. Pro více informací se zeptejte svého lékaře nebo lékárníka.
Zavolejte svého lékaře o radu ohledně nežádoucích účinků. Nežádoucí účinky můžete hlásit úřadu FDA na čísle 1-800-FDA-1088.
POPIS
ZETIA (ezetimib) je ve třídě sloučenin snižujících lipidy, které selektivně inhibují střevní absorpci cholesterolu a příbuzných fytosterolů. Chemický název ezetimibu je 1-(4-fluorfenyl)-3(R)-[3-(4-fluorfenyl)-3(S)-hydroxypropyl]-4(S)-(4-hydroxyfenyl)-2-azetidinon . Empirický vzorec je C24H21F2NO3. Jeho molekulová hmotnost je 409,4 a jeho strukturní vzorec je:
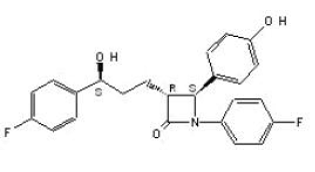
Ezetimib je bílý krystalický prášek, který je snadno až velmi rozpustný v ethanolu, methanolu a acetonu a prakticky nerozpustný ve vodě. Ezetimib má bod tání přibližně 163 °C a je stabilní při teplotě okolí. ZETIA 10 mg je k dispozici jako tableta pro perorální podání obsahující 10 mg ezetimibu a následující neúčinné složky: sodná sůl kroskarmelózy NF, monohydrát laktózy NF, magnesium-stearát NF, mikrokrystalická celulóza NF, povidon USP a laurylsulfát sodný NF.
INDIKACE
Léčba látkami ovlivňujícími lipidy by měla být pouze jednou složkou intervence s více rizikovými faktory u jedinců s významně zvýšeným rizikem aterosklerotického vaskulárního onemocnění v důsledku hypercholesterolémie. Medikamentózní terapie je indikována jako doplněk diety, pokud odpověď na dietu s omezením nasycených tuků a cholesterolu a dalších nefarmakologických opatření samotná byla nedostatečná.
Primární hyperlipidémie
Monoterapie
ZETIA® podávaná samostatně je indikována jako doplňková terapie k dietě ke snížení zvýšeného celkového cholesterolu (celkový-C), cholesterolu v lipoproteinech s nízkou hustotou (LDL-C), apolipoproteinu B (Apo B) a cholesterolu v lipoproteinech s nízkou hustotou. (non-HDL-C) u pacientů s primární (heterozygotní familiární a nefamiliární) hyperlipidemií.
Kombinovaná léčba s inhibitory HMG-CoA reduktázy (statiny)
ZETIA 10 mg podávaná v kombinaci s inhibitorem 3-hydroxy-3-methylglutaryl-koenzym A (HMG-CoA) reduktázy (statinem) je indikována jako doplňková léčba k dietě ke snížení zvýšeného celkového C, LDL-C, Apo B a non-HDL-C u pacientů s primární (heterozygotní familiární a nefamiliární) hyperlipidemií.
Kombinovaná terapie s fenofibrátem
ZETIA 10 mg podávaná v kombinaci s fenofibrátem je indikována jako přídatná léčba k dietě ke snížení zvýšeného celkového cholesterolu, LDL-C, Apo B a non-HDL-C u dospělých pacientů se smíšenou hyperlipidémií.
Homozygotní familiární hypercholesterolémie (HoFH)
Kombinace přípravku ZETIA 10 mg a atorvastatinu nebo simvastatinu je indikována ke snížení zvýšených hladin celkového-C a LDL-C u pacientů s HoFH jako doplněk k jiné hypolipidemické léčbě (např. LDL aferéza) nebo pokud taková léčba není dostupná. .
Homozygotní sitosterolémie
ZETIA 10 mg je indikována jako přídatná léčba k dietě ke snížení zvýšených hladin sitosterolu a kampesterolu u pacientů s homozygotní familiární sitosterolémií.
Omezení použití
Účinek přípravku ZETIA na kardiovaskulární morbiditu a mortalitu nebyl stanoven.
Přípravek ZETIA nebyl studován u Fredricksonových dyslipidémií typu I, III, IV a V.
DÁVKOVÁNÍ A PODÁVÁNÍ
Obecné informace o dávkování
Doporučená dávka přípravku ZETIA je 10 mg jednou denně.
ZETIA 10 mg lze podávat s jídlem nebo bez jídla.
Souběžná terapie snižující hladinu lipidů
ZETIA 10 mg lze podávat se statinem (u pacientů s primární hyperlipidémií) nebo s fenofibrátem (u pacientů se smíšenou hyperlipidémií) pro zvýšení účinku. Pro pohodlí lze denní dávku přípravku ZETIA užívat současně se statinem nebo fenofibrátem podle doporučení pro dávkování pro příslušné léky.
Coadminis se sekvestranty žlučových kyselin
Dávkování přípravku ZETIA by mělo probíhat buď ≥ 2 hodiny před nebo ≥ 4 hodiny po podání sekvestrantu žlučových kyselin (viz DROGOVÉ INTERAKCE ].
Pacienti s poruchou funkce jater
U pacientů s mírnou poruchou funkce jater není nutná žádná úprava dávkování (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Pacienti s poruchou funkce ledvin
U pacientů s poruchou funkce ledvin není nutná žádná úprava dávkování (viz KLINICKÁ FARMAKOLOGIE ]. Při podávání simvastatinu se simvastatinem u pacientů se středně těžkou až těžkou poruchou funkce ledvin (odhadovaná rychlost glomerulární filtrace Použití ve specifických populacích ].
Geriatričtí pacienti
geriatrických pacientů není nutná žádná úprava dávkování (viz KLINICKÁ FARMAKOLOGIE ].
JAK DODÁVÁNO
Dávkové formy A Síly
10mg tablety jsou bílé až téměř bílé tablety ve tvaru tobolky s vyraženým „414“ na jedné straně.
Skladování A Manipulace
č. 3861 - Tablety ZETIA, 10 mg , jsou bílé až téměř bílé tablety ve tvaru tobolky s vyraženým „414“ na jedné straně. Dodávají se následovně:
NDC 66582-414-31 láhve po 30 NDC 66582-414-54 lahví po 90 NDC 66582-414-74 lahví po 500 NDC 66582-414-76 lahví po 5000 NDC 66582-414-28 balení jednotkových dávek po 100 kusech.
Úložný prostor
Skladujte při teplotě 25 °C (77 °F); povoleny výchylky do 15-30°C (59-86°F). [Vidět USP řízená pokojová teplota .] Chraňte před vlhkostí.
Merck Sharp & Dohme Corp., dceřiná společnost MERCK & CO., INC., Whitehouse Station, NJ 08889, USA. Revize: srpen 2013
VEDLEJŠÍ EFEKTY
Následující závažné nežádoucí účinky jsou podrobněji popsány v jiných částech štítku:
- Abnormality jaterních enzymů [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]
- Rabdomyolýza a myopatie [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]
Monoterapeutické studie
databázi kontrolovaných klinických studií ZETIA 10 mg (kontrolovaných placebem) 2396 pacientů s mediánem trvání léčby 12 týdnů (rozmezí 0 až 39 týdnů), 3,3 % pacientů na ZETIA 10 mg a 2,9 % pacientů na placebu přerušilo léčbu kvůli nežádoucím účinkům reakce. Nejčastější nežádoucí účinky ve skupině pacientů léčených přípravkem ZETIA 10 mg, které vedly k přerušení léčby a vyskytovaly se častěji než placebo, byly:
- Artralgie (0,3 %)
- Závrať (0,2 %)
- Zvýšení gama-glutamyltransferázy (0,2 %)
Nejčastěji hlášené nežádoucí účinky (incidence ≥ 2 % a vyšší než u placeba) v databázi kontrolovaných klinických studií ZETIA s monoterapií 2396 pacientů byly: infekce horních cest dýchacích (4,3 %), průjem (4,1 %), artralgie (3,0 %), sinusitida (2,8 %) a bolest končetin (2,7 %).
Studie společného podávání statinů
databázi kontrolovaných klinických studií ZETIA + statin zahrnující 11 308 pacientů s mediánem trvání léčby 8 týdnů (rozmezí 0 až 112 týdnů), 4,0 % pacientů užívajících ZETIA + statin a 3,3 % pacientů užívajících samotný statin kvůli nežádoucím účinkům ukončilo léčbu. Nejčastější nežádoucí účinky ve skupině pacientů léčených přípravkem ZETIA + statin, které vedly k přerušení léčby a vyskytovaly se častěji než samotný statin, byly:
- Zvýšená alaninaminotransferáza (0,6 %)
- Myalgie (0,5 %)
- Únava, zvýšená aspartátaminotransferáza, bolest hlavy a bolest končetin (vždy 0,2 %)
Nejčastěji hlášené nežádoucí účinky (incidence ≥ 2 % a vyšší než samotný statin) v databázi kontrolovaných klinických studií ZETIA + statin u 11 308 pacientů byly: nazofaryngitida (3,7 %), myalgie (3,2 %), infekce horních cest dýchacích (2,9 % ), artralgie (2,6 %) a průjem (2,5 %).
Zkušenosti z klinických studií
Vzhledem k tomu, že klinické studie jsou prováděny za velmi rozdílných podmínek, nelze míry nežádoucích reakcí pozorované v klinických studiích léku přímo srovnávat s mírami v klinických studiích jiného léku a nemusí odrážet míry pozorované v klinické praxi.
Monoterapie
V 10 dvojitě zaslepených, placebem kontrolovaných klinických studiích bylo 2396 pacientů s primární hyperlipidémií (věkové rozmezí 9–86 let, 50 % žen, 90 % bělochů, 5 % černochů, 3 % Hispánců, 2 % Asiatů) a zvýšeným LDL-C byli léčeni přípravkem ZETIA 10 mg/den po střední dobu léčby 12 týdnů (rozmezí 0 až 39 týdnů).
Nežádoucí účinky hlášené u ≥ 2 % pacientů léčených přípravkem ZETIA a s incidencí vyšší než u placeba v placebem kontrolovaných studiích přípravku ZETIA 10 mg, bez ohledu na posouzení kauzality, jsou uvedeny v tabulce 1.
TABULKA 1: Klinické nežádoucí reakce vyskytující se u ≥ 2 % pacientů léčených přípravkem ZETIA 10 mg a s výskytem vyšším než u placeba, bez ohledu na příčinnou souvislost
Frekvence méně častých nežádoucích účinků byla srovnatelná mezi přípravkem ZETIA 10 mg a placebem.
Kombinace se statinem
Ve 28 dvojitě zaslepených, kontrolovaných (placebem nebo aktivně kontrolovaných) klinických studiích bylo 11 308 pacientů s primární hyperlipidémií (věkové rozmezí 10–93 let, 48 % žen, 85 % bělochů, 7 % černochů, 4 % Hispánců, 3 % Asiatů) a zvýšený LDL-C byli léčeni přípravkem ZETIA 10 mg/den souběžně s probíhající statinovou léčbou nebo k ní přidáni po střední dobu léčby 8 týdnů (rozmezí 0 až 112 týdnů).
Incidence po sobě jdoucích zvýšení transamináz (≥3 × ULN) byla vyšší u pacientů užívajících přípravek ZETIA 10 mg podávaný se statiny (1,3 %) než u pacientů léčených samotnými statiny (0,4 %). [Vidět VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]
Klinické nežádoucí účinky hlášené u ≥ 2 % pacientů léčených přípravkem ZETIA + statin a s incidencí vyšší než statin, bez ohledu na posouzení kauzality, jsou uvedeny v tabulce 2.
TABULKA 2: Klinické nežádoucí reakce vyskytující se u ≥ 2 % pacientů léčených přípravkem ZETIA 10 mg podávaným společně se statinem a s výskytem vyšším než statin, bez ohledu na příčinnou souvislost
Kombinace s fenofibrátem
Tato klinická studie zahrnující 625 pacientů se smíšenou dyslipidémií (věkové rozmezí 20–76 let, 44 % žen, 79 % bělochů, 0,1 % černochů, 11 % Hispánců, 5 % Asiatů) léčených po dobu až 12 týdnů a 576 pacientů léčených po dobu až dalších 48 týdnů hodnotilo společné podávání přípravku ZETIA a fenofibrátu. Tato studie nebyla navržena tak, aby porovnávala léčebné skupiny pro méně časté příhody. Incidence (95% CI) klinicky významných zvýšení (≥3 — ULN, konsekutivní) hladin jaterních transamináz byla 4,5 % (1,9, 8,8) a 2,7 % (1,2, 5,4) pro monoterapii fenofibrátem (n=188) a současně užívající přípravek ZETIA s fenofibrátem (n=183), v daném pořadí, upraveným na expozici léčbě. Odpovídající incidence cholecystektomie byla 0,6 % (95% CI: 0,0 %, 3,1 %) a 1,7 % (95% CI: 0,6 %, 4,0 %) pro monoterapii fenofibrátem a ZETIA 10 mg podávaná současně s fenofibrátem, v daném pořadí [viz DROGOVÉ INTERAKCE ]. Počty pacientů vystavených současnému podávání i monoterapii fenofibrátem a ezetimibem nebyly dostatečné pro posouzení rizika onemocnění žlučníku. V žádné z léčebných skupin nedošlo ke zvýšení CPK > 10 — ULN.
Postmarketingové zkušenosti
Vzhledem k tomu, že níže uvedené reakce jsou hlášeny dobrovolně z populace nejisté velikosti, není obecně možné spolehlivě odhadnout jejich frekvenci nebo stanovit příčinnou souvislost s expozicí léku.
Během používání přípravku ZETIA po schválení byly zjištěny následující další nežádoucí účinky:
Hypersenzitivní reakce, včetně anafylaxe, angioedému, vyrážky a kopřivky; erythema multiforme; artralgie; myalgie; zvýšená kreatinfosfokináza; myopatie/rabdomyolýza [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]; zvýšení jaterních transamináz; hepatitida; bolest břicha; trombocytopenie; pankreatitida; nevolnost; závrať; parestézie; Deprese; bolest hlavy; cholelitiáza; cholecystitidu.
DROGOVÉ INTERAKCE
[Vidět KLINICKÁ FARMAKOLOGIE ]
Cyklosporin
Při současném užívání přípravku ZETIA 10 mg a cyklosporinu je třeba opatrnosti z důvodu zvýšené expozice jak ezetimibu, tak cyklosporinu. U pacientů užívajících přípravek ZETIA a cyklosporin by měly být monitorovány koncentrace cyklosporinu.
Stupeň zvýšení expozice ezetimibu může být větší u pacientů s těžkou renální insuficiencí. U pacientů léčených cyklosporinem je třeba pečlivě zvážit potenciální účinky zvýšené expozice ezetimibu ze současného užívání oproti přínosům změn hladin lipidů způsobených ezetimibem.
Fibráty
Účinnost a bezpečnost současného podávání ezetimibu s fibráty jinými než fenofibrát nebyly studovány.
Fibráty mohou zvýšit vylučování cholesterolu do žluči, což vede k cholelitiáze. V předklinické studii u psů ezetimib zvýšil cholesterol ve žlučníku (viz Neklinická toxikologie ]. Současné podávání přípravku ZETIA s fibráty jinými než fenofibrát se nedoporučuje, dokud nebude použití u pacientů náležitě prozkoumáno.
fenofibrát
Při podezření na cholelitiázu u pacienta užívajícího ZETIA 10 mg a fenofibrát jsou indikovány žlučníkové studie a je třeba zvážit alternativní hypolipidemickou léčbu [viz NEŽÁDOUCÍ REAKCE a označení produktu pro fenofibrát].
Cholestyramin
Současné podávání cholestyraminu snížilo průměrnou plochu pod křivkou (AUC) celkového ezetimibu přibližně o 55 %. Přírůstkové snížení LDL-C v důsledku přidání ezetimibu k cholestyraminu může být touto interakcí sníženo.
Kumarinové antikoagulanty
Pokud je ezetimib přidán k warfarinu, kumarinovému antikoagulancii, je třeba náležitě sledovat mezinárodní normalizovaný poměr (INR).
VAROVÁNÍ
Zahrnuto jako součást OPATŘENÍ sekce.
OPATŘENÍ
Používejte se statiny nebo fenofibrátem
Současné podávání přípravku ZETIA se specifickým statinem nebo fenofibrátem by mělo být v souladu s označením produktu pro daný lék.
Jaterní enzymy
kontrolovaných klinických studiích monoterapie byla incidence po sobě jdoucích zvýšení (≥3 x horní hranice normy [ULN]) hladin jaterních transamináz podobná u přípravku ZETIA (0,5 %) a placeba (0,3 %).
V kontrolovaných klinických kombinačních studiích přípravku ZETIA 10 mg zahájených současně se statinem byla incidence po sobě jdoucích zvýšení (≥3 x ULN) hladin jaterních transamináz 1,3 % u pacientů léčených přípravkem ZETIA podávaným se statiny a 0,4 % u pacientů léčených samotnými statiny. Tato zvýšení transamináz byla obecně asymptomatická, nesouvisela s cholestázou a po přerušení léčby nebo při pokračování v léčbě se vrátila na výchozí hodnoty. Pokud je přípravek ZETIA podáván současně se statinem, měly by být na začátku léčby a podle doporučení statinu provedeny jaterní testy. Pokud přetrvává zvýšení ALT nebo AST ≥3 x ULN, zvažte vysazení přípravku ZETIA 10 mg a/nebo statinu.
Myopatie/Rhabdomyolýza
klinických studiích nedošlo k žádné nadměrné myopatii nebo rhabdomyolýze spojené s přípravkem ZETIA 10 mg ve srovnání s relevantním kontrolním ramenem (placebo nebo samotný statin). Nicméně myopatie a rhabdomyolýza jsou známé nežádoucí účinky statinů a jiných léků snižujících lipidy. V klinických studiích byla incidence kreatinfosfokinázy (CPK) > 10 x ULN 0,2 % u přípravku ZETIA vs. 0,1 % u placeba a 0,1 % u přípravku ZETIA podávaného společně se statinem vs. 0,4 % u samotných statinů. Riziko toxicity kosterního svalstva se zvyšuje s vyššími dávkami statinu, vyšším věkem (> 65), hypotyreózou, poruchou funkce ledvin a v závislosti na použitém statinu i při současném užívání jiných léků.
Po uvedení přípravku ZETIA na trh byly hlášeny případy myopatie a rhabdomyolýzy. Většina pacientů, u kterých se rozvinula rhabdomyolýza, užívala statin před zahájením léčby přípravkem ZETIA. Rabdomyolýza však byla hlášena u přípravku ZETIA 10 mg v monoterapii a po přidání přípravku ZETIA 10 mg k látkám, o nichž je známo, že jsou spojeny se zvýšeným rizikem rhabdomyolýzy, jako jsou fibráty. ZETIA a jakýkoli statin nebo fibrát, které pacient současně užívá, by měly být okamžitě vysazeny, pokud je diagnostikována nebo podezření na myopatii. Přítomnost svalových příznaků a hladina CPK > 10 x ULN svědčí pro myopatii.
Poškození jater
Vzhledem k neznámým účinkům zvýšené expozice ezetimibu u pacientů se středně těžkou až těžkou poruchou funkce jater se přípravek ZETIA u těchto pacientů nedoporučuje. [Vidět KLINICKÁ FARMAKOLOGIE ]
Informace pro pacienty
Vidět Označení pacienta schválené FDA (INFORMACE PRO PACIENTA).
Pacienti by měli být poučeni, aby dodržovali doporučenou dietu v Národním programu Cholesterol Education Program (NCEP), pravidelný cvičební program a pravidelné testování lipidového panelu nalačno.
Bolest svalů
Všichni pacienti zahajující léčbu ezetimibem by měli být informováni o riziku myopatie a měli by být poučeni, aby neprodleně hlásili jakoukoli nevysvětlitelnou svalovou bolest, citlivost nebo slabost. Riziko tohoto výskytu se zvyšuje při užívání určitých typů léků. Pacienti by měli všechny léky, a to jak na předpis, tak i volně prodejné, probrat se svým lékařem.
Jaterní enzymy
Jaterní testy by měly být provedeny, pokud je přípravek ZETIA 10 mg přidán k léčbě statiny a v souladu s doporučeními pro statiny.
Těhotenství
Ženám ve fertilním věku je třeba doporučit, aby při užívání přípravku ZETIA přidané k léčbě statiny používaly účinnou metodu antikoncepce k zabránění otěhotnění. Diskutujte se svými pacientkami o budoucích těhotenských plánech a prodiskutujte, kdy ukončit kombinovanou léčbu přípravkem ZETIA 10 mg a statiny, pokud se snaží otěhotnět. Pacientky je třeba upozornit, že pokud otěhotní, měly by přestat užívat kombinovanou léčbu přípravkem ZETIA 10 mg a statiny a zavolat svému lékaři.
Kojení
Kojícím ženám je třeba doporučit, aby neužívaly přípravek ZETIA přidaný k léčbě statiny. Pacientky, které mají poruchu lipidů a kojí, by měly být poučeny, aby prodiskutovaly možnosti se svými zdravotníky.
Neklinická toxikologie
Karcinogeneze, mutageneze, zhoršení plodnosti
Studie karcinogenity ezetimibu trvající 104 týdnů byla provedena na potkanech v dávkách až 1500 mg/kg/den (samci) a 500 mg/kg/den (samice) (~20x vyšší expozice u člověka při dávce 10 mg denně na základě AUC0 -24 hodin pro celkový ezetimib). Studie kancerogenity ezetimibu trvající 104 týdnů byla také provedena na myších v dávkách až 500 mg/kg/den (> 150násobek expozice u člověka při dávce 10 mg denně na základě AUC0-24h pro celkový ezetimib). Nebylo zjištěno žádné statisticky významné zvýšení výskytu nádorů u potkanů nebo myší léčených léčivem.
V testu mikrobiální mutagenity (Ames) se Salmonella typhimurium a Escherichia coli s metabolickou aktivací nebo bez ní nebyly pozorovány žádné známky mutagenity in vitro. V testu chromozomálních aberací v lidských lymfocytech periferní krve s metabolickou aktivací nebo bez ní nebyly pozorovány žádné známky klastogenity in vitro. Kromě toho nebyl v in vivo mikronukleárním testu na myších prokázán žádný důkaz genotoxicity.
Ve studiích fertility ezetimibu po perorálním podání (žaludeční sondou) provedených na potkanech nebyly žádné důkazy reprodukční toxicity při dávkách až 1000 mg/kg/den u samců nebo samic potkanů (~7x vyšší expozice u člověka při dávce 10 mg denně na základě AUC0- 24 hodin pro celkový ezetimib).
Použití u konkrétních populací
Těhotenství
Kategorie těhotenství C
Neexistují žádné adekvátní a dobře kontrolované studie s ezetimibem u těhotných žen. Ezetimib by měl být během těhotenství podáván pouze v případě, že potenciální přínos ospravedlňuje riziko pro plod.
Ve studiích perorálního (žaludeční sondou) embryofetálního vývoje ezetimibu provedených na potkanech a králících během organogeneze nebyly při testovaných dávkách (250, 500, 1000 mg/kg/den) prokázány embryoletální účinky. U potkanů byla při dávce 1000 mg/kg/den (~10násobek expozice u člověka při dávce 10 mg denně na základě AUC0-24h) pozorována zvýšená incidence běžných fetálních kostních nálezů (extra pár hrudních žeber, neosifikovaný střed krčních obratlů, zkrácená žebra) pro celkový ezetimib). U králíků léčených ezetimibem byl pozorován zvýšený výskyt extrahrudních žeber při dávce 1000 mg/kg/den (150x vyšší expozice u člověka při dávce 10 mg denně na základě AUC0-24h pro celkový ezetimib). Ezetimib procházel placentou, když byly březím potkanům a králíkům podávány opakované perorální dávky.
Studie s opakovanými dávkami ezetimibu podávaného v kombinaci se statiny u potkanů a králíků během organogeneze vedly k vyšším expozicím ezetimibu a statinu. Reprodukční nálezy se objevují při nižších dávkách v kombinované terapii ve srovnání s monoterapií.
Všechny statiny jsou kontraindikovány u těhotných a kojících žen. Pokud je přípravek ZETIA 10 mg podáván se statinem ženě ve fertilním věku, podívejte se na kategorii těhotenství a označení statinu. [Viz KONTRAINDIKACE]
Kojící matky
Není známo, zda se ezetimib vylučuje do lidského mateřského mléka. Ve studiích na potkanech byla expozice celkovému ezetimibu u kojených mláďat až poloviční ve srovnání s expozicí pozorovanou v mateřské plazmě. Vzhledem k tomu, že mnoho léků se vylučuje do mateřského mléka, je třeba opatrnosti při podávání přípravku ZETIA 10 mg kojící ženě. Přípravek ZETIA by neměl být podáván kojícím matkám, pokud potenciální přínos neospravedlňuje potenciální riziko pro kojence.
Pediatrické použití
Účinky přípravku ZETIA podávaného se simvastatinem (n=126) ve srovnání s monoterapií simvastatinem (n=122) byly hodnoceny u dospívajících chlapců a dívek s heterozygotní familiární hypercholesterolemií (HeFH). V multicentrické, dvojitě zaslepené, kontrolované studii, po které následovala otevřená fáze, bylo 142 chlapců a 106 postmenarchálních dívek ve věku 10 až 17 let (průměrný věk 14,2 let, 43 % žen, 82 % bělochů, 4 % Asiatů, 2 % černochů , 13 % multirasových) s HeFH byli randomizováni k léčbě buď ZETIA 10 mg společně se simvastatinem nebo simvastatinem v monoterapii. Zařazení do studie vyžadovalo 1) výchozí hladinu LDL-C mezi 160 a 400 mg/dl a 2) anamnézu a klinický obraz v souladu s HeFH. Průměrná výchozí hodnota LDL-C byla 225 mg/dl (rozmezí: 161–351 mg/dl) ve skupině ZETIA 10 mg podávané současně se simvastatinem ve srovnání s 219 mg/dl (rozmezí: 149–336 mg/dl) v monoterapii simvastatinem skupina. Pacienti dostávali souběžně ZETIA a simvastatin (10 mg, 20 mg nebo 40 mg) nebo simvastatin v monoterapii (10 mg, 20 mg nebo 40 mg) po dobu 6 týdnů, současně podávali ZETIA a 40 mg simvastatin nebo 40 mg simvastatin v monoterapii dalších 27 týdnů a poté po dobu 20 týdnů společně podávat ZETIA a simvastatin (10 mg, 20 mg nebo 40 mg).
Výsledky studie v týdnu 6 jsou shrnuty v tabulce 3. Výsledky v týdnu 33 byly konzistentní s výsledky v týdnu 6.
TABULKA 3: Průměrný procentuální rozdíl v týdnu 6 mezi skupinou ZETIA 10 mg podávanou společně se simvastatinem a skupinou souhrnné monoterapie simvastatinem u dospívajících pacientů s heterozygotní familiární hypercholesterolemií
Od začátku studie do konce 33. týdne došlo k přerušení léčby z důvodu nežádoucí reakce u 7 (6 %) pacientů ve skupině ZETIA podávané současně se simvastatinem a u 2 (2 %) pacientů ve skupině s monoterapií simvastatinem.
Během studie se zvýšení jaterních transamináz (dvě po sobě jdoucí měření ALT a/nebo AST ≥3 x ULN) objevilo u čtyř (3 %) jedinců ve skupině ZETIA 10 mg podávané se simvastatinem a u dvou (2 %) jedinců v monoterapii simvastatinem skupina. Zvýšení CPK (≥10 x ULN) se vyskytlo u dvou (2 %) jedinců ve skupině léčené přípravkem ZETIA současně se simvastatinem a u žádného jedince ve skupině s monoterapií simvastatinem.
V této omezené kontrolované studii nebyl zjištěn žádný významný účinek na růst nebo sexuální dospívání u dospívajících chlapců nebo dívek nebo na délku menstruačního cyklu u dívek.
Současné podávání přípravku ZETIA 10 mg se simvastatinem v dávkách vyšších než 40 mg/den nebylo u dospívajících studováno. Přípravek ZETIA 10 mg také nebyl studován u pacientů mladších 10 let nebo u premenarchálních dívek.
Na základě celkového ezetimibu (ezetimib + ezetimib-glukuronid) neexistují žádné farmakokinetické rozdíly mezi dospívajícími a dospělými. Farmakokinetické údaje u pediatrické populace ve věku
Geriatrické použití
Monoterapeutické studie
Z 2396 pacientů, kteří dostávali přípravek ZETIA 10 mg v klinických studiích, bylo 669 (28 %) ve věku 65 let a více a 111 (5 %) bylo ve věku 75 let a více.
Studie společného podávání statinů
Z 11 308 pacientů, kteří v klinických studiích dostávali přípravek ZETIA + statin, bylo 3 587 (32 %) ve věku 65 let a více a 924 (8 %) bylo ve věku 75 let a více.
Mezi těmito pacienty a mladšími pacienty nebyly pozorovány žádné celkové rozdíly v bezpečnosti a účinnosti a další hlášené klinické zkušenosti nezjistily rozdíly v odpovědích mezi staršími a mladšími pacienty, nelze však vyloučit větší citlivost některých starších jedinců [viz KLINICKÁ FARMAKOLOGIE ].
Renální poškození
Při použití jako monoterapie není nutná úprava dávkování přípravku ZETIA 10 mg.
Ve studii Study of Heart and Renal Protection (SHARP) u 9270 pacientů se středně těžkou až těžkou poruchou funkce ledvin (6247 nedialyzovaných pacientů s mediánem sérového kreatininu 2,5 mg/dl a mediánem odhadované glomerulární filtrace 25,6 ml/min/1,73 m² a 3 023 dialyzovaných pacientů), výskyt závažných nežádoucích příhod, nežádoucích příhod vedoucích k přerušení léčby ve studii nebo nežádoucích příhod zvláštního zájmu (muskuloskeletální nežádoucí příhody, abnormality jaterních enzymů, výskyt rakoviny) byl podobný u pacientů, kteří byli kdy léčeni ezetimibem 10 mg plus simvastatin 20 mg (n=4650) nebo placebo (n=4620) během střední doby sledování 4,9 roku. Protože však porucha funkce ledvin je rizikovým faktorem pro myopatii spojenou se statiny, je třeba u pacientů se středně těžkou až těžkou poruchou funkce ledvin používat dávky simvastatinu přesahující 20 mg s opatrností a pečlivým sledováním při současném podávání s přípravkem ZETIA.
Poškození jater
ZETIA se nedoporučuje u pacientů se středně těžkou až těžkou poruchou funkce jater (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ a KLINICKÁ FARMAKOLOGIE ].
ZETIA 10 mg podávaná současně se statinem je kontraindikována u pacientů s aktivním onemocněním jater nebo nevysvětleným přetrvávajícím zvýšením hladin jaterních transamináz (viz KONTRAINDIKACE ; VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ a KLINICKÁ FARMAKOLOGIE ].
PŘEDÁVKOVAT
V klinických studiích bylo podávání ezetimibu v dávce 50 mg/den 15 zdravým subjektům po dobu až 14 dnů, 40 mg/den 18 pacientům s primární hyperlipidémií po dobu až 56 dnů a 40 mg/den 27 pacientům s homozygotní sitosterolemií po dobu 26 týdnů bylo obecně dobře tolerováno. Jedna pacientka s homozygotní sitosterolemií se náhodně předávkovala ezetimibem 120 mg/den po dobu 28 dnů bez hlášených klinických nebo laboratorních nežádoucích účinků.
V případě předávkování je třeba použít symptomatická a podpůrná opatření.
KONTRAINDIKACE
ZETIA je kontraindikována za následujících stavů:
- Kombinace přípravku ZETIA 10 mg se statinem je kontraindikována u pacientů s aktivním onemocněním jater nebo nevysvětlitelným přetrvávajícím zvýšením hladin jaterních transamináz.
- Ženy, které jsou těhotné nebo mohou otěhotnět. Protože statiny snižují syntézu cholesterolu a možná i syntézu dalších biologicky aktivních látek odvozených od cholesterolu, může přípravek ZETIA v kombinaci se statinem způsobit poškození plodu při podávání těhotným ženám. Navíc neexistuje žádný zjevný přínos léčby během těhotenství a bezpečnost u těhotných žen nebyla stanovena. Pokud pacientka otěhotní během užívání tohoto léku, pacientka by měla být seznámena s potenciálním rizikem pro plod a chybějícím známým klinickým přínosem při pokračujícím užívání během těhotenství. [Vidět Použití ve specifických populacích ]
- Kojící matky. Vzhledem k tomu, že statiny mohou přecházet do mateřského mléka a protože statiny mají potenciál způsobit závažné nežádoucí reakce u kojených dětí, ženy, které vyžadují léčbu přípravkem ZETIA v kombinaci se statinem, by měly být poučeny, aby nekojily své děti (viz Použití ve specifických populacích ].
- Pacienti se známou přecitlivělostí na kteroukoli složku tohoto přípravku. U přípravku ZETIA byly hlášeny hypersenzitivní reakce včetně anafylaxe, angioedému, vyrážky a kopřivky (viz NEŽÁDOUCÍ REAKCE ].
KLINICKÁ FARMAKOLOGIE
Mechanismus působení
Ezetimib snižuje hladinu cholesterolu v krvi inhibicí vstřebávání cholesterolu tenkým střevem. Ve dvoutýdenní klinické studii u 18 pacientů s hypercholesterolemií přípravek ZETIA 10 mg inhiboval střevní absorpci cholesterolu o 54 % ve srovnání s placebem. Přípravek ZETIA neměl žádný klinicky významný účinek na plazmatické koncentrace vitamínů A, D a E rozpustných v tucích (ve studii se 113 pacienty) a nezhoršil produkci adrenokortikálních steroidních hormonů (ve studii se 118 pacienty).
Obsah cholesterolu v játrech pochází převážně ze tří zdrojů. Játra mohou syntetizovat cholesterol, přijímat cholesterol z krve z cirkulujících lipoproteinů nebo přijímat cholesterol absorbovaný tenkým střevem. Střevní cholesterol pochází primárně z cholesterolu vylučovaného žlučí a z cholesterolu z potravy.
Ezetimib má mechanismus účinku, který se liší od mechanismu účinku jiných tříd sloučenin snižujících cholesterol (statiny, sekvestranty žlučových kyselin [pryskyřice], deriváty kyseliny fibrové a rostlinné stanoly). Ukázalo se, že molekulárním cílem ezetimibu je přenašeč sterolů, Niemann-Pick C1-Like 1 (NPC1L1), který se podílí na střevním vychytávání cholesterolu a fytosterolů.
Ezetimib neinhibuje syntézu cholesterolu v játrech ani nezvyšuje vylučování žlučových kyselin. Místo toho se ezetimib lokalizuje na kartáčovém okraji tenkého střeva a inhibuje absorpci cholesterolu, což vede ke snížení dodávky střevního cholesterolu do jater. To způsobuje snížení zásob cholesterolu v játrech a zvýšení clearance cholesterolu z krve; tento odlišný mechanismus je komplementární k mechanismu statinů a fenofibrátu [viz Klinické studie ].
Farmakodynamika
Klinické studie prokázaly, že zvýšené hladiny celkového-C, LDL-C a Apo B, hlavní proteinové složky LDL, podporují lidskou aterosklerózu. Snížené hladiny HDL-C jsou navíc spojeny s rozvojem aterosklerózy. Epidemiologické studie prokázaly, že kardiovaskulární morbidita a mortalita se mění přímo s hladinou celkového-C a LDL-C a nepřímo s hladinou HDL-C. Podobně jako LDL mohou aterosklerózu podporovat i lipoproteiny bohaté na triglyceridy bohaté na cholesterol, včetně lipoproteinů s velmi nízkou hustotou (VLDL), lipoproteinů se střední hustotou (IDL) a zbytků. Nezávislý účinek zvýšení HDL-C nebo snížení TG na riziko koronární a kardiovaskulární morbidity a mortality nebyl stanoven.
ZETIA snižuje celkový-C, LDL-C, Apo B, non-HDL-C a TG a zvyšuje HDL-C u pacientů s hyperlipidemií. Podávání přípravku ZETIA 10 mg se statinem je účinné při zlepšení celkového sérového C, LDL-C, Apo B, non-HDL-C, TG a HDL-C nad rámec jedné z těchto léčebných metod. Podávání přípravku ZETIA 10 mg s fenofibrátem je účinné při zlepšení celkového cholesterolu, LDL-C, Apo B a non-HDL-C v séru u pacientů se smíšenou hyperlipidémií ve srovnání s každou léčbou samostatně. Účinky ezetimibu podávaného buď samostatně nebo jako doplněk ke statinu nebo fenofibrátu na kardiovaskulární morbiditu a mortalitu nebyly stanoveny.
Farmakokinetika
Vstřebávání
Po perorálním podání je ezetimib absorbován a rozsáhle konjugován na farmakologicky aktivní fenolický glukuronid (ezetimib-glukuronid). Po jednorázové dávce 10 mg přípravku ZETIA 10 mg dospělým nalačno bylo průměrných maximálních plazmatických koncentrací (Cmax) 3,4 až 5,5 ng/ml dosaženo během 4 až 12 hodin (Tmax). Průměrné hodnoty Cmax ezetimib-glukuronidu 45 až 71 ng/ml byly dosaženy za 1 až 2 hodiny (Tmax). Mezi 5 a 20 mg nebyla žádná podstatná odchylka od proporcionality dávky. Absolutní biologickou dostupnost ezetimibu nelze určit, protože sloučenina je prakticky nerozpustná ve vodném médiu vhodném pro injekci.
Vliv Potraviny Na Orální Absorpci
Současné podávání jídla (s vysokým obsahem tuku nebo bez tuku) nemělo žádný vliv na rozsah absorpce ezetimibu, pokud byl podáván jako ZETIA 10 mg tablety. Hodnota C ezetimibu se zvýšila o 38 % při konzumaci jídel s vysokým obsahem tuku. Přípravek ZETIA lze podávat s jídlem nebo bez jídla.
Rozdělení
Ezetimib a ezetimib-glukuronid se silně váží (> 90 %) na lidské plazmatické proteiny.
Metabolismus A Vylučování
Ezetimib je primárně metabolizován v tenkém střevě a játrech prostřednictvím glukuronidové konjugace (reakce fáze II) s následnou biliární a renální exkrecí. U všech hodnocených druhů byl pozorován minimální oxidativní metabolismus (reakce fáze I).
U lidí je ezetimib rychle metabolizován na ezetimib-glukuronid. Ezetimib a ezetimibeglukuronid jsou hlavními sloučeninami odvozenými od léčiva detekovanými v plazmě a tvoří přibližně 10 až 20 % a 80 až 90 % celkového léčiva v plazmě. Jak ezetimib, tak ezetimib-glukuronid jsou eliminovány z plazmy s poločasem přibližně 22 hodin pro ezetimib i ezetimibeglukuronid. Profily plazmatické koncentrace-čas vykazují mnohonásobné vrcholy, což naznačuje enterohepatální recyklaci.
Po perorálním podání 14C-ezetimibu (20 mg) lidským subjektům představoval celkový ezetimib (ezetimib + ezetimib-glukuronid) přibližně 93 % celkové radioaktivity v plazmě. Po 48 hodinách nebyly v plazmě detekovatelné hladiny radioaktivity.
Přibližně 78 % a 11 % podané radioaktivity bylo nalezeno ve stolici a v moči během 10denního období sběru. Ezetimib byl hlavní složkou ve stolici a představoval 69 % podané dávky, zatímco ezetimib-glukuronid byl hlavní složkou v moči a představoval 9 % podané dávky.
Specifické populace
Geriatričtí pacienti: Ve studii s opakovanými dávkami ezetimibu podávaným 10 mg jednou denně po dobu 10 dnů byly plazmatické koncentrace celkového ezetimibu asi 2krát vyšší u starších (≥65 let) zdravých subjektů ve srovnání s mladšími subjekty.
Pediatričtí pacienti: [Vidět Použití ve specifických populacích ]
Rod: Ve studii s opakovaným podáváním ezetimibu v dávce 10 mg jednou denně po dobu 10 dnů byly plazmatické koncentrace celkového ezetimibu mírně vyšší (
Závod: Na základě metaanalýzy farmakokinetických studií s opakovanými dávkami nebyly zjištěny žádné farmakokinetické rozdíly mezi černými a bělochy. Studie u asijských subjektů ukázaly, že farmakokinetika ezetimibu byla podobná farmakokinetice pozorované u bělochů.
Poškození jater: Po jednorázové dávce 10 mg ezetimibu se průměrná AUC celkového ezetimibu zvýšila přibližně 1,7krát u pacientů s mírnou poruchou funkce jater (Child-Pugh skóre 5 až 6) ve srovnání se zdravými jedinci. Průměrné hodnoty AUC pro celkový ezetimib a ezetimib byly zvýšeny přibližně 3 až 4krát a 5 až 6krát, v tomto pořadí, u pacientů se středně těžkou (Child-Pugh skóre 7 až 9) nebo těžkou poruchou funkce jater (Child-Pugh skóre 10 až 15). Ve 14denní studii s opakovanými dávkami (10 mg denně) u pacientů se středně těžkou poruchou funkce jater byly průměrné hodnoty AUC celkového ezetimibu a ezetimibu zvýšeny přibližně 4krát v den 1 a den 14 ve srovnání se zdravými jedinci. Vzhledem k neznámým účinkům zvýšené expozice ezetimibu u pacientů se středně těžkou nebo těžkou poruchou funkce jater se přípravek ZETIA u těchto pacientů nedoporučuje (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Renální poškození: Po jednorázové dávce 10 mg ezetimibu u pacientů se závažným onemocněním ledvin (n=8; průměr CrCl ≥ 30 ml/min/1,73 m²) byly průměrné hodnoty AUC pro celkový ezetimib, ezetimib-glukuronid a ezetimib zvýšeny přibližně o 1,5 -násobek ve srovnání se zdravými subjekty (n=9).
Lékové interakce
[Viz také DROGOVÉ INTERAKCE ]
ZETIA 10 mg neměl žádný významný účinek na sérii testovacích léků (kofein, dextromethorfan, tolbutamid a IV midazolam), o kterých je známo, že jsou metabolizovány cytochromem P450 (1A2, 2D6, 2C8/9 a 3A4) v „koktejlové“ studii dvanácti zdravých dospělých samců. To naznačuje, že ezetimib není ani inhibitorem ani induktorem těchto izoenzymů cytochromu P450 a je nepravděpodobné, že by ezetimib ovlivňoval metabolismus léků, které jsou těmito enzymy metabolizovány.
TABULKA 4: Účinek současně podávaných léků na celkový ezetimib
TABULKA 5: Účinek současného podávání ezetimibu na systémovou expozici jiným lékům
Toxikologie zvířat a/nebo farmakologie
Hypocholesterolemický účinek ezetimibu byl hodnocen na cholesterolem krmených opicích Rhesus, psech, potkanech a myších modelech metabolismu lidského cholesterolu. Bylo zjištěno, že ezetimib má hodnotu ED50 0,5 μg/kg/den pro inhibici vzestupu plazmatických hladin cholesterolu u opic. Hodnoty ED50 u psů, potkanů a myší byly 7, 30 a 700 μg/kg/den, v daném pořadí. Tyto výsledky jsou v souladu s tím, že ZETIA je silným inhibitorem absorpce cholesterolu.
Na potkaním modelu, kde byl glukuronidový metabolit ezetimibu (SCH 60663) podáván intraduodenálně, byl metabolit stejně účinný jako mateřská sloučenina (SCH 58235) při inhibici vstřebávání cholesterolu, což naznačuje, že glukuronidový metabolit měl aktivitu podobnou mateřské lék.
1měsíčních studiích u psů, kterým byl podáván ezetimib (0,03 až 300 mg/kg/den), se koncentrace cholesterolu ve žlučníku zvýšila ~2- až 4násobně. Nicméně dávka 300 mg/kg/den podávaná psům po dobu jednoho roku nevedla k tvorbě žlučových kamenů ani k jiným nepříznivým hepatobiliárním účinkům. Ve 14denní studii na myších, kterým byl podáván ezetimib (0,3 až 5 mg/kg/den) a krmeny nízkotučnou dietou nebo dietou bohatou na cholesterol, nebyla koncentrace cholesterolu ve žlučové žluči buď ovlivněna, nebo byla snížena na normální hodnoty.
Byla provedena řada akutních předklinických studií ke stanovení selektivity přípravku ZETIA pro inhibici absorpce cholesterolu. Ezetimib inhiboval absorpci 14C-cholesterolu bez vlivu na absorpci triglyceridů, mastných kyselin, žlučových kyselin, progesteronu, ethinylestradiolu nebo vitamínů A a D rozpustných v tucích.
Ve 4- až 12týdenních studiích toxicity u myší ezetimib neindukoval enzymy metabolizující léky cytochromu P450. Ve studiích toxicity byla pozorována farmakokinetická interakce ezetimibu se statiny (rodiči nebo jejich aktivní hydroxykyselinové metabolity) u potkanů, psů a králíků.
Klinické studie
Primární hyperlipidémie
ZETIA 10 mg snižuje celkový-C, LDL-C, Apo B, non-HDL-C a TG a zvyšuje HDL-C u pacientů s hyperlipidemií. Maximální až téměř maximální odpovědi je obecně dosaženo během 2 týdnů a udržuje se během chronické terapie.
Monoterapie
Ve dvou multicentrických, dvojitě zaslepených, placebem kontrolovaných, 12týdenních studiích u 1719 pacientů s primární hyperlipidémií ZETIA významně snížila celkový-C, LDL-C, Apo B, non-HDL-C a TG a zvýšila HDL- C ve srovnání s placebem (viz tabulka 6). Snížení LDL-C bylo konzistentní napříč věkem, pohlavím a výchozím LDL-C.
TABULKA 6: Odpověď na ZETIA 10 mg u pacientů s primární hyperlipidémií (průměrná % změna oproti neléčené výchozí hodnotě†)
Kombinace se statiny
ZETIA 10 mg přidána k průběžné statinové terapii
V multicentrické, dvojitě zaslepené, placebem kontrolované, 8týdenní studii se zúčastnilo 769 pacientů s primární hyperlipidémií, známým koronárním srdečním onemocněním nebo více kardiovaskulárními rizikovými faktory, kteří již dostávali statiny v monoterapii, ale kteří nesplnili svůj cíl NCEP ATP II LDL Cíl -C byli randomizováni tak, aby dostávali buď ZETIA 10 mg nebo placebo navíc k jejich pokračujícímu statinu.
ZETIA 10 mg, přidaná k probíhající léčbě statiny, významně snížila celkový-C, LDL-C, Apo B, non-HDL-C a TG a zvýšila HDL-C ve srovnání se statinem podávaným samostatně (viz tabulka 7). Snížení LDL-C vyvolané přípravkem ZETIA 10 mg bylo obecně konzistentní u všech statinů.
TABULKA 7: Reakce na přidání ZETIA 10 mg k průběžné statinové terapii u pacientů s hyperlipidémií (průměrná % změna od léčené výchozí hodnoty‡)
ZETIA zahájena současně se statinem
Ve čtyřech multicentrických, dvojitě zaslepených, placebem kontrolovaných, 12týdenních studiích byl u 2382 hyperlipidemických pacientů podáván přípravek ZETIA nebo placebo samostatně nebo s různými dávkami atorvastatinu, simvastatinu, pravastatinu nebo lovastatinu.
Když byli všichni pacienti užívající ZETIA se statinem porovnáni se všemi pacienty užívajícími odpovídající statin samotný, ZETIA 10 mg významně snížila celkový-C, LDL-C, Apo B, non-HDL-C a TG, a s výjimkou pravastatinu zvýšení HDL-C ve srovnání se statinem podávaným samostatně. Snížení LDL-C vyvolané přípravkem ZETIA 10 mg bylo obecně konzistentní u všech statinů. (Viz poznámku pod čarou, tabulky 8 až 11.)
TABULKA 8: Odpověď na ZETIA 10 mg a atorvastatin zahájená souběžně u pacientů s primární hyperlipidémií (průměrná % změna oproti neléčené výchozí hodnotě†)
TABULKA 9: Odpověď na ZETIA 10 mg a simvastatin zahájená souběžně u pacientů s primární hyperlipidémií (průměrná % změna oproti neléčené výchozí hodnotě†)
TABULKA 10: Odpověď na léčbu ZETIA a pravastatin zahájenou souběžně u pacientů s primární hyperlipidémií (průměrná % změna oproti neléčené výchozí hodnotě†)
TABULKA 11: Odpověď na léčbu ZETIA a lovastatin zahájenou souběžně u pacientů s primární hyperlipidémií (průměrná % změna oproti neléčené výchozí hodnotě†)
Kombinace s fenofibrátem
V multicentrické, dvojitě zaslepené, placebem kontrolované klinické studii u pacientů se smíšenou hyperlipidémií bylo 625 pacientů léčeno po dobu až 12 týdnů a 576 pacientů po dobu až dalších 48 týdnů. Ve 12týdenní studii byli pacienti randomizováni k užívání placeba, samotného přípravku ZETIA, samotného fenofibrátu v dávce 160 mg nebo fenofibrátu v dávce 10 mg a 160 mg přípravku ZETIA. Po dokončení 12týdenní studie byli způsobilí pacienti zařazeni do skupiny ZETIA podávané společně s fenofibrátem nebo monoterapií fenofibrátem po dobu dalších 48 týdnů.
ZETIA 10 mg podávaná současně s fenofibrátem významně snížila celkový C, LDL-C, Apo B a non-HDL-C ve srovnání s fenofibrátem podávaným samostatně. Procentuální snížení TG a procentuální zvýšení HDLC pro přípravek ZETIA podávaný současně s fenofibrátem byly srovnatelné s těmi u fenofibrátu podávaného samostatně (viz tabulka 12).
TABULKA 12: Odpověď na ZETIA 10 mg a fenofibrát zahájená souběžně u pacientů se smíšenou hyperlipidémií (průměrná % změna oproti neléčené výchozí hodnotě† ve 12. týdnu†)
Změny v koncových bodech lipidů po dalších 48 týdnech léčby přípravkem ZETIA podávaným společně s fenofibrátem nebo s fenofibrátem samotným byly v souladu s 12týdenními údaji uvedenými výše.
Homozygotní familiární hypercholesterolémie (HoFH)
Byla provedena studie s cílem posoudit účinnost přípravku ZETIA při léčbě HoFH. Tato dvojitě zaslepená, randomizovaná, 12týdenní studie zahrnovala 50 pacientů s klinickou a/nebo genotypovou diagnózou HoFH, se současnou LDL aferézou nebo bez ní, kteří již užívali atorvastatin nebo simvastatin (40 mg). Pacienti byli randomizováni do jedné ze tří léčebných skupin, atorvastatin nebo simvastatin (80 mg), ZETIA 10 mg podávaná s atorvastatinem nebo simvastatinem (40 mg) nebo ZETIA podávaná s atorvastatinem nebo simvastatinem (80 mg). Vzhledem ke snížené biologické dostupnosti ezetimibu u pacientů současně užívajících cholestyramin (viz DROGOVÉ INTERAKCE ], byl ezetimib podáván nejméně 4 hodiny před nebo po podání pryskyřic. Průměrná výchozí hodnota LDL-C byla 341 mg/dl u pacientů randomizovaných na atorvastatin 80 mg nebo simvastatin 80 mg samotný a 316 mg/dl ve skupině randomizované na ZETIA 10 mg plus atorvastatin 40 nebo 80 mg nebo simvastatin 40 nebo 80 mg. ZETIA 10 mg podávaná s atorvastatinem nebo simvastatinem (skupiny se statiny 40 a 80 mg, souhrnně) významně snížila LDL-C (21 %) ve srovnání se zvýšením dávky simvastatinu nebo atorvastatinu v monoterapii ze 40 na 80 mg (7 %). U pacientů léčených přípravkem ZETIA 10 mg plus 80 mg atorvastatinu nebo přípravkem ZETIA 10 mg plus 80 mg simvastatinu se LDL-C snížil o 27 %.
Homozygotní sitosterolémie (fytosterolémie)
Byla provedena studie s cílem posoudit účinnost přípravku ZETIA při léčbě homozygotní sitosterolémie. V této multicentrické, dvojitě zaslepené, placebem kontrolované, 8týdenní studii se zúčastnilo 37 pacientů s homozygotní sitosterolémií se zvýšenými hladinami sitosterolu v plazmě (> 5 mg/dl) na jejich současném terapeutickém režimu (dieta, pryskyřice vázající žlučové kyseliny, statiny, operace ileálního bypassu a/nebo LDL aferéza), byli randomizováni k léčbě ZETIA (n=30) nebo placeba (n=7). Vzhledem ke snížené biologické dostupnosti ezetimibu u pacientů současně užívajících cholestyramin (viz DROGOVÉ INTERAKCE ], byl ezetimib podáván alespoň 2 hodiny před nebo 4 hodiny po podání pryskyřic. S výjimkou jednoho subjektu, který dostával LDL aferézu, přípravek ZETIA významně snížil plazmatický sitosterol o 21 % a kampesterol o 24 % oproti výchozí hodnotě. Naproti tomu u pacientů, kteří dostávali placebo, došlo ke zvýšení sitosterolu o 4 % a kampesterolu o 3 % oproti výchozí hodnotě. U pacientů léčených přípravkem ZETIA 10 mg se průměrné plazmatické hladiny rostlinných sterolů v průběhu studie progresivně snižovaly. Účinky snížení plazmatického sitosterolu a kampesterolu na snížení rizik kardiovaskulární morbidity a mortality nebyly stanoveny.
Snížení sitosterolu a kampesterolu bylo konzistentní mezi pacienty užívajícími přípravek ZETIA 10 mg současně se sekvestranty žlučových kyselin (n=8) a pacienty, kteří nebyli současně léčeni sekvestranty žlučových kyselin (n=21).
Omezení použití
Účinek přípravku ZETIA 10 mg na kardiovaskulární morbiditu a mortalitu nebyl stanoven.
INFORMACE PRO PACIENTA
ZETIA® (ezetimib) Tablety
Informace pro pacienty o ZETIA (zet´-ea)
Generický název: ezetimib (e-zet´-e-mib)
Přečtěte si pozorně tyto informace dříve, než začnete užívat přípravek ZETIA® a pokaždé, když dostanete více přípravku ZETIA. Mohou se objevit nové informace. Tyto informace nenahrazují rozhovor s lékařem o vašem zdravotním stavu nebo léčbě. Máte-li jakékoli dotazy týkající se přípravku ZETIA, zeptejte se svého lékaře. Pouze váš lékař může určit, zda je pro vás přípravek ZETIA vhodný.
Co je ZETIA?
ZETIA je lék používaný ke snížení hladiny celkového cholesterolu a LDL (špatného) cholesterolu v krvi. Přípravek ZETIA je určen pro pacienty, kteří nemohou kontrolovat hladinu cholesterolu pouze dietou a cvičením. Může se používat samostatně nebo s jinými léky k léčbě vysokého cholesterolu. Během užívání tohoto léku byste měli zůstat na dietě snižující cholesterol.
ZETIA působí tak, že snižuje množství cholesterolu, které vaše tělo absorbuje. ZETIA 10 mg vám nepomůže zhubnout. Nebylo prokázáno, že přípravek ZETIA 10 mg zabraňuje srdečním onemocněním nebo srdečním záchvatům.
Další informace o cholesterolu naleznete v části „Co bych měl vědět o vysokém cholesterolu?“ oddíl, který následuje.
Kdo by neměl užívat ZETIA 10 mg?
- Neužívejte přípravek ZETIA 10 mg, pokud jste alergický(á) na ezetimib, léčivou látku přípravku ZETIA, nebo na neúčinné složky. Seznam neaktivních složek naleznete v následující části „Neaktivní složky“.
- Pokud trpíte aktivním onemocněním jater, neužívejte přípravek ZETIA 10 mg při užívání léků na snížení cholesterolu nazývaných statiny.
- Pokud jste těhotná nebo kojíte, neužívejte přípravek ZETIA při užívání statinu.
- Jste-li žena v plodném věku, měla byste používat účinnou metodu antikoncepce k zabránění otěhotnění při užívání přípravku ZETIA přidané k léčbě statiny.
Přípravek ZETIA nebyl studován u dětí mladších 10 let.
Co mám říci svému lékaři před a během užívání přípravku ZETIA 10 mg?
Informujte svého lékaře o všech lécích na předpis a bez předpisu, které užíváte nebo plánujete užívat, včetně přírodních nebo rostlinných přípravků.
Informujte svého lékaře o všech svých zdravotních potížích včetně alergií.
Informujte svého lékaře, pokud:
- měl někdy problémy s játry. ZETIA 10 mg pro vás nemusí být to pravé.
- jste těhotná nebo plánujete otěhotnět. Váš lékař s vámi prodiskutuje, zda je pro vás přípravek ZETIA 10 mg vhodný.
- kojí. Nevíme, zda se ZETIA 10 mg může dostat k vašemu dítěti prostřednictvím vašeho mléka. Váš lékař s vámi prodiskutuje, zda je pro vás přípravek ZETIA 10 mg vhodný.
- pociťovat nevysvětlitelné bolesti svalů, citlivost nebo slabost.
Jak mám užívat ZETIA 10 mg?
- Užívejte přípravek ZETIA jednou denně, s jídlem nebo bez jídla. Může být snazší zapamatovat si dávku, pokud ji užíváte každý den ve stejnou dobu, například se snídaní, večeří nebo před spaním. Pokud užíváte také jiný lék na snížení cholesterolu, zeptejte se svého lékaře, zda je můžete užívat současně.
- Jestliže jste zapomněl(a) užít přípravek ZETIA, vezměte si ji, jakmile si vzpomenete. Neužívejte však více než jednu dávku přípravku ZETIA denně.
- Během užívání přípravku ZETIA pokračujte v dodržování diety na snížení cholesterolu. Zeptejte se svého lékaře, pokud potřebujete informace o dietě.
- Pokračujte v užívání přípravku ZETIA 10 mg, dokud Vám lékař neřekne, abyste přestala. Je důležité, abyste pokračoval(a) v užívání přípravku ZETIA 10 mg, i když se necítíte špatně.
- Pravidelně navštěvujte svého lékaře, aby vám zkontroloval hladinu cholesterolu a zkontroloval nežádoucí účinky. Před zahájením užívání přípravku ZETIA se statinem a během léčby vám může lékař provést krevní testy, aby zkontroloval vaše játra.
Jaké jsou možné vedlejší účinky přípravku ZETIA?
klinických studiích pacienti při užívání přípravku ZETIA hlásili málo nežádoucích účinků. Ty zahrnovaly průjem, bolesti kloubů a pocit únavy.
Pacienti zaznamenali závažné svalové problémy při užívání přípravku ZETIA 10 mg, obvykle když byl přípravek ZETIA 10 mg přidán ke statinovému léku. Pokud se u Vás během užívání přípravku ZETIA objeví nevysvětlitelná bolest svalů, citlivost nebo slabost, okamžitě kontaktujte svého lékaře. Musíte to udělat okamžitě, protože ve vzácných případech mohou být tyto svalové problémy vážné, s rozpadem svalů vedoucím k poškození ledvin.
Kromě toho byly při běžném používání hlášeny následující nežádoucí účinky: alergické reakce (které mohou vyžadovat okamžitou léčbu) včetně otoků obličeje, rtů, jazyka a/nebo hrdla, které mohou způsobit potíže s dýcháním nebo polykáním, vyrážka a kopřivka ; vystouplá červená vyrážka, někdy s terčovitými lézemi; bolest kloubů; Bolest svalů; změny v některých laboratorních krevních testech; problémy s játry; bolest břicha; zánět slinivky břišní; nevolnost; závrať; pocit mravenčení; Deprese; bolest hlavy; žlučové kameny; zánětu žlučníku.
Informujte svého lékaře, pokud máte tyto nebo jiné zdravotní problémy během léčby přípravkem ZETIA. Úplný seznam nežádoucích účinků si vyžádejte od svého lékaře nebo lékárníka.
Co bych měl vědět o vysokém cholesterolu?
Cholesterol je druh tuku, který se nachází ve vaší krvi. Váš celkový cholesterol se skládá z LDL a HDL cholesterolu.
LDL cholesterol se nazývá „špatný“ cholesterol, protože se může hromadit ve stěně vašich tepen a vytvářet plaky. Postupem času může hromadění plaku způsobit zúžení tepen. Toto zúžení může zpomalit nebo blokovat průtok krve do vašeho srdce, mozku a dalších orgánů. Vysoký LDL cholesterol je hlavní příčinou srdečních onemocnění a jednou z příčin mrtvice.
HDL cholesterol se nazývá „dobrý“ cholesterol, protože zabraňuje hromadění špatného cholesterolu v tepnách.
Triglyceridy jsou také tuky, které se nacházejí ve vaší krvi.
Obecné informace o společnosti ZETIA
Léky jsou někdy předepisovány na stavy, které nejsou uvedeny v příbalových informacích pro pacienty. Neužívejte přípravek ZETIA 10 mg při onemocnění, pro které nebyl předepsán. Nedávejte přípravek ZETIA jiným lidem, i když mají stejný stav jako vy. Může jim to ublížit.
Zde jsou shrnuty nejdůležitější informace o společnosti ZETIA. Pokud chcete více informací, promluvte si se svým lékařem. Můžete požádat svého lékárníka nebo lékaře o informace o přípravku ZETIA 10 mg, které jsou určeny pro zdravotníky.
Neaktivní složky:
Sodná sůl kroskarmelózy, monohydrát laktózy, magnesium-stearát, mikrokrystalická celulóza, povidon a laurylsulfát sodný.