Xeloda 500mg Capecitabine Použití, vedlejší účinky a dávkování. Cena v internetové lékárně. Generické léky bez předpisu.
Co je Xeloda 500 mg a jak se používá?
Xeloda je lék na předpis používaný k léčbě příznaků rakoviny, jako je rakovina tlustého střeva, kolorektální rakovina a rakovina prsu. Xeloda lze užívat samostatně nebo s jinými léky.
Xeloda patří do třídy léků nazývaných Antineoplastika, Antimetabolit.
Není známo, zda je přípravek Xeloda 500 mg bezpečný a účinný u dětí.
Jaké jsou možné nežádoucí účinky přípravku Xeloda 500 mg?
Xeloda může způsobit závažné nežádoucí účinky včetně:
- horečka nad 100,5 stupňů,
- nevolnost,
- ztráta chuti k jídlu,
- jíst mnohem méně než obvykle,
- zvracení (více než jednou za 24 hodin),
- těžký průjem (více než 4krát denně nebo v noci),
- puchýře nebo vředy v ústech,
- červené nebo oteklé dásně,
- potíže s polykáním,
- bolest, citlivost, zarudnutí, otok, puchýře nebo olupování kůže na rukou nebo nohou,
- pocit velké žízně nebo horka,
- neschopnost močit,
- silné pocení,
- horká a suchá pokožka,
- bolest nebo tlak na hrudi,
- nerovnoměrné srdeční tepy,
- dušnost,
- otok nebo rychlý nárůst hmotnosti,
- bolestivé nebo obtížné močení,
- otoky nohou nebo kotníků,
- cítit se unaveně,
- dušnost,
- tmavá moč,
- stolice hliněné barvy,
- zežloutnutí kůže nebo očí (žloutenka),
- horečka nebo jiné příznaky chřipky,
- kašel,
- kožní vředy,
- bledá kůže,
- snadná tvorba modřin,
- neobvyklé krvácení,
- pocit točení hlavy,
- rychlý srdeční tep,
- bolest krku,
- otok v obličeji nebo na jazyku,
- pálení v očích a
- bolest kůže, po níž následuje červená nebo fialová vyrážka (zejména v obličeji nebo horní části těla) a způsobuje puchýře a olupování
Pokud máte některý z výše uvedených příznaků, okamžitě vyhledejte lékařskou pomoc.
Mezi nejčastější nežádoucí účinky přípravku Xeloda 500 mg patří:
- bolest břicha,
- zácpa,
- žaludeční nevolnost,
- pocit únavy,
- mírná kožní vyrážka a
- necitlivost nebo mravenčení v rukou nebo nohou
Informujte lékaře, pokud máte jakýkoli nežádoucí účinek, který vás obtěžuje nebo který neustupuje.
Toto nejsou všechny možné vedlejší účinky přípravku Xeloda. Pro více informací se zeptejte svého lékaře nebo lékárníka.
Zavolejte svého lékaře o radu ohledně nežádoucích účinků. Nežádoucí účinky můžete hlásit úřadu FDA na čísle 1-800-FDA-1088.
VAROVÁNÍ
INTERAKCE XELODA-WARFARIN
Interakce XELODA 500 mg warfarinu: U pacientů, kteří dostávají souběžně kapecitabin a perorální antikoagulační léčbu deriváty kumarinu, by měla být často monitorována antikoagulační odpověď (INR nebo protrombinový čas), aby bylo možné odpovídajícím způsobem upravit dávku antikoagulancia. Klinicky významná léková interakce XELODA-Warfarin byla prokázána v klinické farmakologické studii [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ DROGOVÉ INTERAKCE]. U pacientů užívajících XELODA 500 mg současně s antikoagulancii kumarinovými deriváty, jako je warfarin a fenprokumon, byly hlášeny změněné koagulační parametry a/nebo krvácení, včetně úmrtí. Postmarketingové zprávy ukázaly klinicky významné zvýšení protrombinového času (PT) a INR u pacientů, kteří byli v době zavedení přípravku XELODA 500 mg stabilizováni na antikoagulanciích. Tyto příhody se objevily během několika dnů a až několika měsíců po zahájení léčby přípravkem XELODA 500 mg a v několika případech během 1 měsíce po ukončení léčby přípravkem XELODA. Tyto příhody se vyskytly u pacientů s jaterními metastázami i bez nich. Věk vyšší než 60 let a diagnóza rakoviny nezávisle předurčují pacienty ke zvýšenému riziku koagulopatie.
POPIS
XELODA (kapecitabin) je fluoropyrimidinkarbamát s antineoplastickou aktivitou. Jedná se o perorálně podávané systémové proléčivo 5'-deoxy-5-fluoruridin (5'-DFUR), které se přeměňuje na 5-fluoruracil.
Chemický název kapecitabinu je 5'-deoxy-5-fluor-N-[(pentyloxy)karbonyl]-cytidin a má molekulovou hmotnost 359,35. Kapecitabin má následující strukturní vzorec:
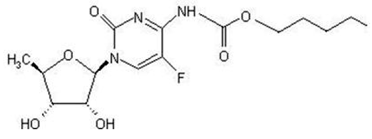
Kapecitabin je bílý až téměř bílý krystalický prášek s rozpustností ve vodě 26 mg/ml při 20 °C.
XELODA se dodává jako bikonvexní, podlouhlé potahované tablety pro perorální podání. Jedna světle broskvová tableta obsahuje 150 mg kapecitabinu a jedna broskvově zbarvená tableta obsahuje 500 mg kapecitabinu. Mezi neaktivní složky přípravku XELODA patří: bezvodá laktóza, sodná sůl kroskarmelózy, hydroxypropylmethylcelulóza, mikrokrystalická celulóza, magnesium-stearát a čištěná voda. Broskvový nebo světle broskvový filmový povlak obsahuje hydroxypropylmethylcelulózu, mastek, oxid titaničitý a syntetické žluté a červené oxidy železa.
INDIKACE
Kolorektální karcinom
- XELODA je indikována jako jediná látka k adjuvantní léčbě u pacientů s Dukesovým C karcinomem tlustého střeva, kteří podstoupili kompletní resekci primárního nádoru, když je preferována léčba samotným fluoropyrimidinem. XELODA 500 mg nebyla horší než 5-fluorouracil a leukovorin (5-FU/LV) pro přežití bez onemocnění (DFS). Lékaři by měli vzít v úvahu výsledky studií kombinované chemoterapie, které prokázaly zlepšení DFS a OS, když předepisují monoterapii XELODA 500 mg v adjuvantní léčbě Dukesova C karcinomu tlustého střeva.
- XELODA 500 mg je indikována jako léčba první volby u pacientů s metastatickým kolorektálním karcinomem, kdy je preferována léčba samotným fluoropyrimidinem. Kombinovaná chemoterapie prokázala přínos v přežití ve srovnání se samotnou 5-FU/LV. Přínos přežití oproti 5-FU/LV nebyl u XELODA 500 mg v monoterapii prokázán. Použití XELODA místo 5FU/LV v kombinacích nebylo dostatečně studováno, aby byla zajištěna bezpečnost nebo zachování výhody přežití.
Rakovina prsu
- XELODA v kombinaci s docetaxelem je indikována k léčbě pacientek s metastatickým karcinomem prsu po selhání předchozí chemoterapie obsahující antracykliny.
- XELODA 500 mg v monoterapii je také indikována k léčbě pacientek s metastatickým karcinomem prsu rezistentních na paklitaxel i chemoterapeutický režim obsahující antracykliny nebo rezistentních na paclitaxel au kterých není indikována další léčba antracykliny (např. pacientky, které dostaly kumulativní dávky 400 mg/m2 doxorubicinu nebo ekvivalentů doxorubicinu). Rezistence je definována jako progresivní onemocnění během léčby, s počáteční odpovědí nebo bez ní, nebo jako relaps do 6 měsíců po dokončení léčby adjuvantním režimem obsahujícím antracykliny.
DÁVKOVÁNÍ A PODÁVÁNÍ
Důležité pokyny pro administraci
Tablety XELODA se mají spolknout celé a zapít vodou do 30 minut po jídle. XELODA 500 mg je cytotoxický lék. Dodržujte příslušné speciální postupy pro manipulaci a likvidaci.1 Pokud je nutné tablety XELODA 500 mg rozřezat nebo rozdrtit, měl by to provést odborník vyškolený v bezpečné manipulaci s cytotoxickými léky za použití vhodného vybavení a bezpečnostních postupů. Dávka XELODA se vypočítá podle plochy povrchu těla.
Standardní počáteční dávka
Monoterapie (metastatický kolorektální karcinom, adjuvantní kolorektální karcinom, metastatický karcinom prsu)
Doporučená dávka XELODA je 1250 mg/m2 podávaná perorálně dvakrát denně (ráno a večer; ekvivalentní celkové denní dávce 2500 mg/m2) po dobu 2 týdnů, po nichž následuje 1 týdenní přestávka podávaná ve 3týdenních cyklech (viz ).
Adjuvantní léčba u pacientů s Dukesovým karcinomem C tlustého střeva se doporučuje po dobu celkem 6 měsíců [tj. XELODA 1250 mg/m2 perorálně dvakrát denně po dobu 2 týdnů, po nichž následuje 1 týdenní přestávka, podávaná jako 3týdenní cykly pro celkovou 8 cyklů (24 týdnů)].
V kombinaci s docetaxelem (metastatický karcinom prsu)
V kombinaci s docetaxelem je doporučená dávka XELODA 1 250 mg/m2 dvakrát denně po dobu 2 týdnů, po nichž následuje 1 týdenní přestávka, v kombinaci s docetaxelem v dávce 75 mg/m2 ve formě 1hodinové intravenózní infuze každé 3 týdny. Premedikace, podle označení docetaxelu, by měla být zahájena před podáním docetaxelu u pacientů užívajících kombinaci XELODA plus docetaxel. zobrazuje celkovou denní dávku XELODA podle plochy povrchu těla a počtu tablet, které je třeba užít v každé dávce.
Pokyny pro řízení dávky
Všeobecné
Dávkování XELODA může být nutné individualizovat, aby se optimalizovala léčba pacienta. Pacienti by měli být pečlivě sledováni z hlediska toxicity a dávky přípravku XELODA 500 mg by měly být podle potřeby upraveny tak, aby vyhovovaly individuální toleranci pacienta k léčbě (viz Klinické studie ]. Toxicitu způsobenou podáním XELODA 500 mg lze zvládnout symptomatickou léčbou, přerušením dávkování a úpravou dávky XELODA. Jakmile byla dávka snížena, neměla by se později zvyšovat. Dávky XELODA 500 mg vynechané z důvodu toxicity se nenahrazují ani neobnovují; místo toho by měl pacient pokračovat v plánovaných léčebných cyklech.
Dávka fenytoinu a dávka kumarinových derivátů antikoagulancií může být nutné snížit, pokud se kterýkoli lék podává současně s XELODA (viz DROGOVÉ INTERAKCE ].
Monoterapie (metastatický kolorektální karcinom, adjuvantní kolorektální karcinom, metastatický karcinom prsu)
Pro zvládnutí nežádoucích účinků se doporučuje schéma úpravy dávky XELODA popsané níže (viz ).
V kombinaci s docetaxelem (metastatický karcinom prsu)
Úprava dávky XELODA 500 mg kvůli toxicitě by měla být provedena podle výše uvedeného pro XELODA. Pokud je na začátku léčebného cyklu indikováno odložení léčby buď pro XELODA 500 mg nebo docetaxel, pak by mělo být podání obou látek odloženo, dokud nebudou splněny požadavky na opětovné zahájení léčby oběma léky.
Schéma snižování dávky docetaxelu při použití v kombinaci s přípravkem XELODA k léčbě metastatického karcinomu prsu je uvedeno v tabulce 3.
Úprava počáteční dávky u zvláštních populací
Renální poškození
U pacientů s mírnou poruchou funkce ledvin (clearance kreatininu = 51 až 80 ml/min [Cockroft a Gault, jak je uvedeno níže] se nedoporučuje žádná úprava počáteční dávky XELODA 500 mg). U pacientů se středně těžkou poruchou funkce ledvin (výchozí clearance kreatininu = 30 až 50 ml/min) snížení dávky na 75 % počáteční dávky XELODA, pokud se používá jako monoterapie nebo v kombinaci s docetaxelem (z 1250 mg/m2 na 950 mg/m2 dvakrát denně) se doporučuje [viz Použití u konkrétních populací a KLINICKÁ FARMAKOLOGIE ]. Následná úprava dávky se doporučuje, jak je uvedeno v a (v závislosti na režimu), pokud se u pacienta rozvine nežádoucí příhoda stupně 2 až 4 [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]. Doporučení pro úpravu počáteční dávky pro pacienty se středně těžkou poruchou funkce ledvin platí jak pro XELODA 500 mg v monoterapii, tak pro XELODA 500 mg v kombinaci s docetaxelem.
Cockroftova a Gaultova rovnice:
Geriatrie
Lékaři by měli být opatrní při sledování účinků přípravku XELODA 500 mg u starších pacientů. Pro doporučení dávkování nejsou k dispozici dostatečné údaje.
JAK DODÁVÁNO
Síla lékových forem
XELODA se dodává jako bikonvexní, podlouhlé potahované tablety pro perorální podání. Jedna světle broskvová tableta obsahuje 150 mg kapecitabinu a jedna broskvově zbarvená tableta obsahuje 500 mg kapecitabinu.
150 mg
Barva: Světle broskvová Rytina: XELODA na jedné straně a 150 na druhé straně 150 mg tablety jsou baleny v lahvičkách po 60 ( NDC 0004-1100-20), jednotlivě balené v kartonu.
500 mg
Barva: Broskvová Rytina: XELODA 500 mg na jedné straně a 500 mg na druhé straně 500 mg tablety jsou baleny v lahvičkách po 120 ( NDC 0004-1101-50), jednotlivě balené v kartonu.
Skladování A Manipulace
Skladujte při teplotě 25 °C (77 °F); povoleny výchylky do 15° až 30°C (59° až 86°F). [Viz USP Regulovaná pokojová teplota]. UCHOVÁVEJTE TĚSNĚ UZAVŘENÉ.
XELODA je cytotoxický lék. Dodržujte příslušné speciální postupy pro manipulaci a likvidaci.1 Jakýkoli nepoužitý přípravek by měl být zlikvidován v souladu s místními požadavky nebo programy zpětného odběru léků.
REFERENCE
1. „Nebezpečné drogy OSHA.“ OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html.
Distribuuje: Genentech USA, Inc. Člen skupiny Roche Group, 1 DNA Way, South San Francisco, CA 94080-4990. Upraveno: květen 2021
VEDLEJŠÍ EFEKTY
Vzhledem k tomu, že klinické studie jsou prováděny za velmi odlišných podmínek, nelze míry nežádoucích reakcí pozorované v klinických studiích léku přímo srovnávat s mírami v klinických studiích jiného léku a nemusí odrážet míry pozorované v praxi.
Adjuvantní rakovina tlustého střeva
ukazuje nežádoucí reakce vyskytující se u ≥ 5 % pacientů z jedné studie fáze 3 u pacientů s Dukesovou C rakovinou tlustého střeva, kteří dostali alespoň jednu dávku studovaného léku a měli alespoň jedno hodnocení bezpečnosti. Celkem 995 pacientů bylo léčeno 1250 mg/m2 dvakrát denně XELODA 500 mg podávanou po dobu 2 týdnů, po nichž následoval 1 týden bez přestávky, a 974 pacientům byl podáván 5-FU a leukovorin (20 mg/m2 leukovorin IV a následně 425 mg/m2 IV bolus 5FU ve dnech 1-5 každých 28 dní). Medián trvání léčby byl 164 dnů u pacientů léčených kapecitabinem a 145 dnů u pacientů léčených 5-FU/LV. Celkem 112 (11 %) a 73 (7 %) pacientů léčených kapecitabinem a 5-FU/LV léčbu ukončilo kvůli nežádoucím účinkům. Celkem 18 úmrtí ze všech příčin se vyskytlo buď ve studii, nebo do 28 dnů po podání studovaného léku: 8 (0,8 %) pacientů randomizovaných na XELODA 500 mg a 10 (1,0 %) randomizovaných na 5-FU/LV.
ukazuje laboratorní abnormality stupně 3/4 vyskytující se u ≥1 % pacientů z jedné studie fáze 3 u pacientů s Dukesovou C rakovinou tlustého střeva, kteří dostali alespoň jednu dávku studovaného léku a měli alespoň jedno hodnocení bezpečnosti.
Metastatický kolorektální karcinom
Monoterapie
ukazuje nežádoucí účinky vyskytující se u ≥ 5 % pacientů ze spojení dvou studií fáze 3 u metastatického kolorektálního karcinomu první linie. Celkem 596 pacientů s metastatickým kolorektálním karcinomem bylo léčeno 1250 mg/m2 dvakrát denně XELODA podávanou po dobu 2 týdnů s následnou týdenní přestávkou a 593 pacientům byl podáván 5-FU a leukovorin v režimu Mayo (20 mg/m2 leukovorinu IV a následně 425 mg/m2 IV bolus 5-FU, ve dnech 1-5, každých 28 dnů). Ve sloučené kolorektální databázi byl medián trvání léčby 139 dnů u pacientů léčených kapecitabinem a 140 dnů u pacientů léčených 5-FU/LV. Celkem 78 (13 %) a 63 (11 %) pacientů léčených kapecitabinem a 5-FU/LV přerušilo léčbu z důvodu nežádoucích účinků/interkurentního onemocnění. Celkem 82 úmrtí ze všech příčin se vyskytlo buď ve studii, nebo do 28 dnů po podání studovaného léku: 50 (8,4 %) pacientů randomizovaných na XELODA 500 mg a 32 (5,4 %) randomizovaných na 5-FU/LV.
Rakovina prsu
V kombinaci s docetaxelem
Následující údaje jsou uvedeny pro kombinovanou studii s XELODA 500 mg a docetaxelem u pacientek s metastatickým karcinomem prsu v a . V rameni s kombinací XELODA a docetaxelu byla léčba XELODA podávána perorálně 1250 mg/m2 dvakrát denně jako intermitentní léčba (2 týdny léčby následované 1 týdnem bez léčby) po dobu nejméně 6 týdnů a docetaxel podávaný jako 1hodinová intravenózní infuze dávku 75 mg/m2 první den každého 3týdenního cyklu po dobu nejméně 6 týdnů. V rameni s monoterapií byl docetaxel podáván jako 1hodinová intravenózní infuze v dávce 100 mg/m2 první den každého 3týdenního cyklu po dobu nejméně 6 týdnů. Průměrná délka léčby byla 129 dnů v kombinované větvi a 98 dnů v monoterapii. Celkem 66 pacientů (26 %) v rameni s kombinovanou léčbou a 49 (19 %) v rameni s monoterapií odstoupilo ze studie kvůli nežádoucím účinkům. Procento pacientů vyžadujících snížení dávky z důvodu nežádoucích účinků bylo 65 % ve větvi s kombinovanou léčbou a 36 % ve větvi s monoterapií. Procento pacientů vyžadujících přerušení léčby z důvodu nežádoucích účinků v rameni s kombinací bylo 79 %. Přerušení léčby byla součástí schématu úpravy dávky pro rameno s kombinovanou terapií, ale ne pro pacienty léčené docetaxelem v monoterapii.
Monoterapie
Následující údaje jsou uvedeny pro studii u pacientek s rakovinou prsu stadia IV, které dostávaly dávku 1250 mg/m2 podávanou dvakrát denně po dobu 2 týdnů, po které následovala 1 týdenní přestávka. Průměrná délka léčby byla 114 dní. Celkem 13 ze 162 pacientů (8 %) přerušilo léčbu z důvodu nežádoucích účinků/interkurentního onemocnění.
Klinicky relevantní nežádoucí účinky u
Klinicky relevantní nežádoucí účinky hlášené u
Monoterapie (metastatický kolorektální karcinom, adjuvantní kolorektální karcinom, metastatický karcinom prsu)
Gastrointestinální: abdominální distenze, dysfagie, proktalgie, ascites (0,1 %), žaludeční vřed (0,1 %), ileus (0,3 %), toxická dilatace střeva, gastroenteritida (0,1 %)
Kůže a podkoží: porucha nehtů (0,1 %), zvýšené pocení (0,1 %), fotosenzitivní reakce (0,1 %), kožní ulcerace, pruritus, syndrom z ozáření (0,2 %)
Všeobecné: bolest na hrudi (0,2 %), onemocnění podobné chřipce, návaly horka, bolest (0,1 %), chrapot, podrážděnost, potíže s chůzí, žízeň, hmota na hrudi, kolaps, fibróza (0,1 %), krvácení, edém, sedace
Neurologický: nespavost, ataxie (0,5 %), třes, dysfázie, encefalopatie (0,1 %), abnormální koordinace, dysartrie, ztráta vědomí (0,2 %), porucha rovnováhy
Metabolismus: zvýšená hmotnost, kachexie (0,4 %), hypertriglyceridémie (0,1 %), hypokalémie, hypomagnezémie
Oko: zánět spojivek
Respirační: kašel (0,1 %), epistaxe (0,1 %), astma (0,2 %), hemoptýza, respirační tíseň (0,1 %), dušnost
Srdeční: tachykardie (0,1 %), bradykardie, fibrilace síní, ventrikulární extrasystoly, extrasystoly, myokarditida (0,1 %), perikardiální výpotek
Infekce: laryngitida (1,0 %), bronchitida (0,2 %), pneumonie (0,2 %), bronchopneumonie (0,2 %), keratokonjunktivitida, sepse (0,3 %), plísňové infekce (včetně kandidózy) (0,2 %)
Muskuloskeletální: myalgie, bolest kostí (0,1 %), artritida (0,1 %), svalová slabost
Krev a lymfatické: leukopenie (0,2 %), porucha koagulace (0,1 %), útlum kostní dřeně (0,1 %), idiopatická trombocytopenie purpura (1,0 %), pancytopenie (0,1 %)
Cévní: hypotenze (0,2 %), hypertenze (0,1 %), lymfedém (0,1 %), plicní embolie (0,2 %), cévní mozková příhoda (0,1 %)
psychiatrické: deprese, zmatenost (0,1 %)
Renální: poškození ledvin (0,6 %)
Ucho: závrať
Hepatobiliární: jaterní fibróza (0,1 %), hepatitida (0,1 %), cholestatická hepatitida (0,1 %), abnormální jaterní testy
Imunitní systém: přecitlivělost na léky (0,1 %)
XELODA 500 mg v kombinaci s docetaxelem (metastatický karcinom prsu)
Gastrointestinální: ileus (0,4 %), nekrotizující enterokolitida (0,4 %), jícnový vřed (0,4 %), hemoragický průjem (0,8 %)
Neurologický: ataxie (0,4 %), synkopa (1,2 %), ztráta chuti (0,8 %), polyneuropatie (0,4 %), migréna (0,4 %)
Srdeční: supraventrikulární tachykardie (0,4 %)
Infekce: neutropenická sepse (2,4 %), sepse (0,4 %), bronchopneumonie (0,4 %) Krev a
Lymfatický: agranulocytóza (0,4 %), pokles protrombinu (0,4 %)
Cévní: hypotenze (1,2 %), žilní flebitida a tromboflebitida (0,4 %), posturální hypotenze (0,8 %)
Renální: selhání ledvin (0,4 %)
Hepatobiliární: žloutenka (0,4 %), abnormální jaterní testy (0,4 %), jaterní selhání (0,4 %), jaterní kóma (0,4 %), hepatotoxicita (0,4 %)
Imunitní systém: přecitlivělost (1,2 %)
Postmarketingové zkušenosti
Následující nežádoucí účinky byly pozorovány po uvedení na trh: angioedém, jaterní selhání, stenóza slzných cest, akutní selhání ledvin sekundární k dehydrataci včetně fatálního konce (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ], kožní lupus erythematodes, poruchy rohovky včetně keratitidy, toxická leukoencefalopatie, závažné kožní reakce, jako je Stevens-Johnsonův syndrom a toxická epidermální nekrolýza (TEN) [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ], přetrvávající nebo těžký syndrom ruky a nohy může nakonec vést ke ztrátě otisků prstů [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]
V případech expozice rozdrceným tabletám XELODA 500 mg byly hlášeny následující nežádoucí účinky: podráždění a otok očí, kožní vyrážka, průjem, parestézie, bolest hlavy, podráždění žaludku, zvracení a nevolnost.
DROGOVÉ INTERAKCE
Lékové interakce
Antikoagulancia
U pacientů užívajících XELODA 500 mg současně s antikoagulancii kumarinovými deriváty, jako je warfarin a fenprokumon, byly hlášeny změněné koagulační parametry a/nebo krvácení (viz BOX VAROVÁNÍ ]. Tyto příhody se objevily během několika dnů a až několika měsíců po zahájení léčby přípravkem XELODA 500 mg a v několika případech během 1 měsíce po ukončení léčby přípravkem XELODA. Tyto příhody se vyskytly u pacientů s jaterními metastázami i bez nich. Ve studii lékových interakcí s podáním jedné dávky warfarinu došlo k významnému zvýšení průměrné AUC S-warfarinu [viz KLINICKÁ FARMAKOLOGIE ]. Maximální pozorovaná hodnota INR se zvýšila o 91 %. Tato interakce je pravděpodobně způsobena inhibicí cytochromu P450 2C9 kapecitabinem a/nebo jeho metabolity.
fenytoin
Hladina fenytoinu by měla být pečlivě sledována u pacientů užívajících XELODA 500 mg a může být nutné snížit dávku fenytoinu (viz DÁVKOVÁNÍ A PODÁVÁNÍ ]. Postmarketingové zprávy naznačují, že někteří pacienti užívající XELODA 500 mg a fenytoin měli toxicitu spojenou se zvýšenými hladinami fenytoinu. Formální studie lékových interakcí s fenytoinem nebyly provedeny, ale předpokládá se, že mechanismem interakce je inhibice izoenzymu CYP2C9 kapecitabinem a/nebo jeho metabolity.
Leukovorin
Koncentrace 5-fluorouracilu je zvýšená a jeho toxicita může být zvýšena leukovorinem. U starších pacientů užívajících jednou týdně leukovorin a fluorouracil byla hlášena úmrtí na těžkou enterokolitidu, průjem a dehydrataci.
substráty CYP2C9
Kromě warfarinu nebyly provedeny žádné formální studie lékových interakcí mezi XELODA a jinými substráty CYP2C9. Při současném podávání XELODA se substráty CYP2C9 je třeba postupovat opatrně.
allopurinol
Současné užívání s alopurinolem může snížit koncentraci aktivních metabolitů kapecitabinu (viz KLINICKÁ FARMAKOLOGIE ], což může snížit účinnost XELODA. Během léčby přípravkem XELODA se vyvarujte užívání alopurinolu.
Interakce droga-jídlo
Bylo prokázáno, že jídlo snižuje jak rychlost, tak rozsah absorpce kapecitabinu [viz KLINICKÁ FARMAKOLOGIE ]. Ve všech klinických studiích byli pacienti instruováni, aby podávali XELODA 500 mg do 30 minut po jídle. Doporučuje se podávat XELODA s jídlem [viz DÁVKOVÁNÍ A PODÁVÁNÍ ].
VAROVÁNÍ
Zahrnuto jako součást "OPATŘENÍ" Sekce
OPATŘENÍ
Koagulopatie
pacientů, kteří dostávají souběžně kapecitabin a perorální antikoagulační léčbu kumarinovými deriváty, by měla být jejich antikoagulační odpověď (INR nebo protrombinový čas) pečlivě sledována s velkou frekvencí a dávka antikoagulancia by měla být odpovídajícím způsobem upravena [viz BOX VAROVÁNÍ a DROGOVÉ INTERAKCE ].
Průjem
XELODA 500 mg může vyvolat průjem, někdy těžký. Pacienti se závažným průjmem by měli být pečlivě sledováni a v případě dehydratace jim musí být poskytnuty náhrady tekutin a elektrolytů. U 875 pacientů s metastatickým karcinomem prsu nebo kolorektálním karcinomem, kteří dostávali XELODA v monoterapii, byla střední doba do prvního výskytu průjmu 2. až 4. stupně 34 dnů (rozmezí od 1 do 369 dnů). Medián trvání průjmu 3. až 4. stupně byl 5 dní. Průjem 2. stupně National Cancer Institute of Canada (NCIC) je definován jako zvýšení o 4 až 6 stolic/den nebo noční stolice, průjem 3. stupně jako zvýšení o 7 až 9 stolic/den nebo inkontinence a malabsorpce a průjem 4. stupně jako zvýšení o ≥ 10 stolic/den nebo silně krvavý průjem nebo potřeba parenterální podpory. Pokud se objeví průjem 2., 3. nebo 4. stupně, podávání přípravku XELODA by mělo být okamžitě přerušeno, dokud průjem neustoupí nebo se jeho intenzita nesníží na stupeň 1 [viz DÁVKOVÁNÍ A PODÁVÁNÍ ]. Doporučuje se standardní protiprůjmová léčba (např. loperamid).
Byla hlášena nekrotizující enterokolitida (tyflitida).
Kardiotoxicita
Kardiotoxicita pozorovaná u XELODA 500 mg zahrnuje infarkt myokardu/ischémii, anginu pectoris, dysrytmie, srdeční zástavu, srdeční selhání, náhlou smrt, elektrokardiografické změny a kardiomyopatii. Tyto nežádoucí účinky mohou být častější u pacientů s onemocněním koronárních tepen v anamnéze.
Deficit dihydropyrimidindehydrogenázy
Na základě postmarketingových zpráv jsou pacienti s určitými homozygotními nebo určitými složenými heterozygotními mutacemi v genu DPD, které vedou k úplné nebo téměř úplné absenci aktivity DPD, vystaveni zvýšenému riziku akutního raného nástupu toxicity a závažných, život ohrožujících nebo smrtelných nežádoucích účinků. reakce způsobené XELODA (např. mukozitida, průjem, neutropenie a neurotoxicita). Pacienti s částečnou aktivitou DPD mohou mít také zvýšené riziko závažných, život ohrožujících nebo fatálních nežádoucích účinků způsobených přípravkem XELODA.
Ukončete nebo trvale ukončete léčbu přípravkem XELODA 500 mg na základě klinického posouzení nástupu, trvání a závažnosti pozorovaných toxicit u pacientů s prokázanou akutní ranou nebo neobvykle závažnou toxicitou, která může naznačovat téměř úplnou nebo úplnou absenci aktivity DPD. Žádná dávka XELODA nebyla prokázána jako bezpečná pro pacienty s úplnou absencí aktivity DPD. Nejsou k dispozici dostatečné údaje pro doporučení konkrétní dávky u pacientů s částečnou aktivitou DPD měřenou jakýmkoli specifickým testem.
Dehydratace a selhání ledvin
Byla pozorována dehydratace, která může způsobit akutní selhání ledvin, které může být fatální. Vyšší riziko mají pacienti s již existující sníženou funkcí ledvin nebo pacienti, kteří současně užívají XELODA se známými nefrotoxickými látkami. Pacienti s anorexií, astenií, nevolností, zvracením nebo průjmem se mohou rychle dehydratovat. Při podávání přípravku XELODA 500 mg sledujte pacienty, abyste zabránili dehydrataci a napravili ji na jejím počátku. Pokud dojde k dehydrataci 2. (nebo vyššího) stupně, je třeba léčbu XELODOU okamžitě přerušit a dehydrataci upravit. Léčba by neměla být znovu zahájena, dokud není pacient rehydratován a všechny vyvolávající příčiny nebyly opraveny nebo pod kontrolou. Úprava dávky by měla být podle potřeby aplikována pro vyvolávající nežádoucí příhodu [viz DÁVKOVÁNÍ A PODÁVÁNÍ ].
Pacienti se středně těžkou poruchou funkce ledvin na začátku vyžadují snížení dávky (viz DÁVKOVÁNÍ A PODÁVÁNÍ ]. Pacienti s mírnou a středně závažnou poruchou funkce ledvin na začátku léčby by měli být pečlivě sledováni kvůli nežádoucím účinkům. Pokud se u pacienta rozvine nežádoucí příhoda stupně 2 až 4, jak je uvedeno v [viz. DÁVKOVÁNÍ A PODÁVÁNÍ , Použití u konkrétních populací , a KLINICKÁ FARMAKOLOGIE ].
Embryo-fetální toxicita
Na základě zjištění z reprodukčních studií na zvířatech a mechanismu účinku může XELODA způsobit poškození plodu, když je podávána těhotné ženě [viz KLINICKÁ FARMAKOLOGIE ]. Omezené dostupné údaje nejsou dostatečné k informování o použití přípravku XELODA 500 mg u těhotných žen. V reprodukčních studiích na zvířatech způsobilo podávání kapecitabinu březím zvířatům během období organogeneze embryoletalitu a teratogenitu u myší a embryoletalitu u opic při 0,2 a 0,6násobku expozice (AUC) u pacientů užívajících doporučenou dávku [viz Použití u konkrétních populací ]. Informujte těhotné ženy o potenciálním riziku pro plod. Informujte ženy o reprodukčním potenciálu, aby během léčby a po dobu 6 měsíců po poslední dávce přípravku XELODA používaly účinnou antikoncepci (viz Použití u konkrétních populací ].
Mukokutánní A Dermatologická Toxicita
U pacientů léčených přípravkem XELODA se mohou objevit závažné mukokutánní reakce, některé s fatálním koncem, jako je Stevens-Johnsonův syndrom a toxická epidermální nekrolýza (TEN). NEŽÁDOUCÍ REAKCE ]. Léčba přípravkem XELODA 500 mg by měla být trvale ukončena u pacientů, u kterých se vyskytla závažná mukokutánní reakce, kterou lze pravděpodobně připsat léčbě přípravkem XELODA.
Syndrom ruky a nohy (palmárně-plantární erytrodysestezie nebo akrální erytém vyvolaný chemoterapií) je kožní toxicita. Medián doby do nástupu byl 79 dnů (rozmezí od 11 do 360 dnů) s rozsahem závažnosti 1 až 3 u pacientů, kteří dostávali XELODA 500 mg v monoterapii u metastatického onemocnění. Stupeň 1 je charakterizován některým z následujících projevů: necitlivost, dysestézie/parestézie, mravenčení, nebolestivé otoky nebo erytém rukou a/nebo nohou a/nebo nepohodlí, které nenarušuje běžné činnosti. Syndrom ruky a nohy 2. stupně je definován jako bolestivý erytém a otoky rukou a/nebo nohou a/nebo nepohodlí ovlivňující pacientovy aktivity každodenního života. Syndrom ruky a nohy 3. stupně je definován jako vlhká deskvamace, ulcerace, puchýře nebo silná bolest rukou a/nebo nohou a/nebo silné nepohodlí, které způsobuje, že pacient není schopen pracovat nebo vykonávat činnosti každodenního života. Přetrvávající nebo závažný syndrom ruka-noha (stupeň 2 a vyšší) může nakonec vést ke ztrátě otisků prstů, což by mohlo ovlivnit identifikaci pacienta. Pokud se objeví syndrom ruka-noha 2. nebo 3. stupně, podávání XELODA by mělo být přerušeno, dokud příhoda nevymizí nebo se její intenzita nesníží na stupeň 1. Po syndromu ruka-noha 3. stupně by měly být následné dávky XELODA 500 mg sníženy. vidět DÁVKOVÁNÍ A PODÁVÁNÍ ].
Hyperbilirubinémie
875 pacientů s metastatickým karcinomem prsu nebo kolorektálním karcinomem, kteří dostávali alespoň jednu dávku XELODA 1250 mg/m2 dvakrát denně v monoterapii po dobu 2 týdnů s následnou 1 týdenní přestávkou, se objevila hyperbilirubinémie 3. stupně (1,5–3 x ULN). 15,2 % (n=133) pacientů a hyperbilirubinémie 4. stupně (>3 x ULN) se vyskytla u 3,9 % (n=34) pacientů. Z 566 pacientů, kteří měli metastázy v játrech na začátku studie a 309 pacientů bez metastáz v játrech na začátku studie, se hyperbilirubinémie stupně 3 nebo 4 vyskytla u 22,8 % a 12,3 %. Ze 167 pacientů s hyperbilirubinémií stupně 3 nebo 4 mělo 18,6 % (n=31) také postbaseline zvýšení (1. až 4. stupeň, bez zvýšení na začátku) alkalické fosfatázy a 27,5 % (n=46) mělo postbaseline zvýšení transamináz v kdykoli (ne nutně souběžně). Většina těchto pacientů, 64,5 % (n=20) a 71,7 % (n=33), měla na začátku studie jaterní metastázy. Navíc 57,5 % (n = 96) a 35,3 % (n = 59) ze 167 pacientů mělo zvýšení (stupeň 1 až 4) jak před výchozí hodnotou, tak po výchozí hodnotě v alkalické fosfatáze, respektive transaminázách. Pouze 7,8 % (n=13) a 3,0 % (n=5) mělo zvýšení alkalické fosfatázy nebo transamináz 3. nebo 4. stupně.
596 pacientů léčených přípravkem XELODA 500 mg v první linii léčby metastatického kolorektálního karcinomu byla incidence hyperbilirubinémie 3. nebo 4. stupně podobná jako v celkové databázi bezpečnosti přípravku XELODA 500 mg v monoterapii v klinické studii. Medián doby do nástupu hyperbilirubinémie stupně 3 nebo 4 u populace s kolorektálním karcinomem byl 64 dní a medián celkového bilirubinu se zvýšil z 8 μm/l na začátku na 13 μm/l během léčby přípravkem XELODA. Ze 136 pacientů s kolorektálním karcinomem s hyperbilirubinémií stupně 3 nebo 4 mělo 49 pacientů hyperbilirubinémii stupně 3 nebo 4 jako poslední naměřenou hodnotu, z nichž 46 mělo na začátku studie metastázy v játrech.
U 251 pacientek s metastatickým karcinomem prsu, které dostávaly kombinaci XELODA 500 mg a docetaxelu, se hyperbilirubinémie 3. stupně (1,5 až 3 x ULN) vyskytla u 7 % (n=17) a hyperbilirubinémie 4. stupně (> 3 x ULN) u 2 % (n=5).
Pokud se objeví zvýšení bilirubinu stupně 3 až 4 související s lékem, podávání XELODA 500 mg by mělo být okamžitě přerušeno, dokud hyperbilirubinémie neklesne na ≤ 3,0 X ULN (viz doporučené úpravy dávky pod DÁVKOVÁNÍ A PODÁVÁNÍ ].
Hematologické
U 875 pacientů s metastatickým karcinomem prsu nebo kolorektálním karcinomem, kteří dostávali dávku 1250 mg/m2 podávanou dvakrát denně jako monoterapii po dobu 2 týdnů s následnou týdenní přestávkou, mělo 3,2 %, 1,7 % a 2,4 % pacientů 3. nebo 4 neutropenie, trombocytopenie nebo pokles hemoglobinu, v daném pořadí. U 251 pacientek s metastatickým karcinomem prsu, které dostávaly dávku XELODA v kombinaci s docetaxelem, mělo 68 % neutropenii 3. nebo 4. stupně, 2,8 % mělo trombocytopenii 3. nebo 4. stupně a 9,6 % mělo anémii 3. nebo 4. stupně.
Pacienti s výchozím počtem neutrofilů
Geriatričtí pacienti
pacientů ve věku ≥ 80 let může být vyšší výskyt nežádoucích účinků 3. nebo 4. stupně. U 875 pacientů s metastatickým karcinomem prsu nebo kolorektálním karcinomem, kteří dostávali XELODA 500 mg v monoterapii, se u 62 % z 21 pacientů ve věku ≥ 80 let léčených přípravkem XELODA 500 mg objevila nežádoucí příhoda 3. nebo 4. stupně související s léčbou: průjem u 6 (28,6 %) , nauzea u 3 (14,3 %), syndrom ruky a nohy u 3 (14,3 %) a zvracení u 2 (9,5 %) pacientů. Mezi 10 pacienty ve věku 70 let a více (žádní pacienti nebyli starší 80 let) léčených přípravkem XELODA 500 mg v kombinaci s docetaxelem se u 30 % (3 z 10) pacientů vyskytl průjem a stomatitida stupně 3 nebo 4 a 40 % (4 z 10) mělo syndrom ruka-noha 3. stupně.
Mezi 67 pacienty ve věku ≥ 60 let, kteří dostávali XELODA v kombinaci s docetaxelem, výskyt nežádoucích účinků 3. nebo 4. stupně souvisejících s léčbou, závažných nežádoucích účinků souvisejících s léčbou, vysazení kvůli nežádoucím účinkům, přerušení léčby z důvodu nežádoucích účinků a léčby přerušení během prvních dvou léčebných cyklů byla vyšší než ve skupině pacientů
995 pacientů užívajících XELODA jako adjuvantní léčbu Dukesova C karcinomu tlustého střeva po resekci primárního nádoru se u 41 % z 398 pacientů ve věku ≥ 65 let léčených přípravkem XELODA vyskytla nežádoucí příhoda 3. nebo 4. stupně související s léčbou: ruka-a syndrom nohy u 75 (18,8 %), průjem u 52 (13,1 %), stomatitida u 12 (3,0 %), neutropenie/granulocytopenie u 11 (2,8 %), zvracení u 6 (1,5 %) a nauzea u 5 (1,3 %) % pacientů. U pacientů ve věku ≥ 65 let (všechna randomizovaná populace; kapecitabin 188 pacientů, 5-FU/LV 208 pacientů) léčených pro Dukesův C karcinom tlustého střeva po resekci primárního nádoru, poměry rizik pro přežití bez onemocnění a celkové přežití pro XELODA ve srovnání s 5-FU/LV byla 1,01 (95% CI 0,80 – 1,27) a 1,04 (95% CI 0,79 – 1,37).
Jaterní nedostatečnost
Pacienti s mírnou až středně těžkou jaterní dysfunkcí způsobenou jaterními metastázami by měli být při podávání XELODA pečlivě sledováni. Účinek těžké jaterní dysfunkce na dispozici XELODA není znám (viz Použití u konkrétních populací a KLINICKÁ FARMAKOLOGIE ].
Kombinace s jinými drogami
Použití XELODA v kombinaci s irinotekanem nebylo dostatečně studováno.
Informace pro pacienty
Doporučte pacientovi, aby si přečetl označení pacienta schválené FDA ( INFORMACE PRO PACIENTA ).
Průjem
Informujte pacienty, kteří trpí průjmem 2. stupně (zvýšení o 4 až 6 stolic/den nebo noční stolice) nebo vyšším nebo kteří trpí těžkým krvavým průjmem se silnou bolestí břicha a horečkou, aby přestali užívat přípravek XELODA. Informujte pacienty o použití protiprůjmové léčby (např. loperamid) ke zvládnutí průjmu (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Kardiotoxicita
Informujte pacienty o riziku kardiotoxicity a okamžitě kontaktujte svého poskytovatele zdravotní péče nebo jděte na pohotovost kvůli novému výskytu bolesti na hrudi, dušnosti, závrati nebo točení hlavy [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Deficit dihydropyrimidindehydrogenázy
Informujte pacienty, aby informovali svého poskytovatele zdravotní péče, pokud mají známý nedostatek DPD. Informujte pacienty, pokud mají úplnou nebo téměř úplnou absenci aktivity DPD, jsou vystaveni zvýšenému riziku akutního raného nástupu toxicity a závažných, život ohrožujících nebo fatálních nežádoucích reakcí způsobených XELODA (např. mukozitida, průjem, neutropenie a neurotoxicita) [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Dehydratace a selhání ledvin
Poučte pacienty, kteří trpí dehydratací 2. nebo vyššího stupně (indikace IV tekutin VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Důležité pokyny pro administraci
Doporučte pacientům, aby polykali tablety XELODA 500 mg celé a zapíjeli vodou do 30 minut po jídle. Informujte pacienty a pečovatele, aby tablety XELODA nedrtily ani nekrájely. Informujte pacienty, pokud nemohou polykat tablety XELODA celé, aby informovali svého poskytovatele zdravotní péče [viz DÁVKOVÁNÍ A PODÁVÁNÍ ].
Hypersenzitivita a angioedém
Informujte pacienty, že XELODA může způsobit závažné reakce přecitlivělosti a angioedém. Informujte pacienty, kteří mají známou přecitlivělost na kapecitabin nebo 5-fluorouracil, aby informovali svého poskytovatele zdravotní péče [viz KONTRAINDIKACE ]. Poučte pacienty, u kterých se rozvinou reakce přecitlivělosti nebo mukokutánní příznaky (např. kopřivka, vyrážka, erytém, pruritus nebo otok obličeje, rtů, jazyka nebo hrdla, které ztěžují polykání nebo dýchání), aby přestali užívat přípravek XELODA a okamžitě kontaktovali svého poskytovatele zdravotní péče. nebo jít na pohotovost. [vidět NEŽÁDOUCÍ REAKCE ].
Nevolnost
Poučte pacienty, kteří trpí nevolností 2. stupně (příjem potravy výrazně snížený, ale mohou jíst přerušovaně) nebo vyšším, aby okamžitě přestali užívat přípravek XELODA 500 mg a aby kontaktovali svého poskytovatele zdravotní péče o zvládání nevolnosti [viz NEŽÁDOUCÍ REAKCE ].

Zvracení
Poučte pacienty se zvracením stupně 2 (2 až 5 epizod za 24 hodin) nebo vyšším, aby okamžitě přestali užívat přípravek XELODA a aby kontaktovali svého poskytovatele zdravotní péče pro zvládnutí zvracení [viz NEŽÁDOUCÍ REAKCE ].
Syndrom ruky a nohy
Poučte pacienty se syndromem rukou a nohou 2. stupně (bolestivý erytém a otoky rukou a/nebo nohou a/nebo nepohodlí ovlivňující každodenní aktivity pacientů) nebo vyšším, aby okamžitě přestali užívat přípravek XELODA 500 mg a kontaktovali svého poskytovatele zdravotní péče. . Informujte pacienty, že se doporučuje zahájit symptomatickou léčbu a syndrom ruka-noha může vést ke ztrátě otisků prstů, což by mohlo ovlivnit osobní identifikaci [viz NEŽÁDOUCÍ REAKCE ].
Stomatitida
Informujte pacienty se stomatitidou stupně 2 (bolestivý erytém, otok nebo vředy v ústech nebo jazyku, ale mohou jíst) nebo vyšší, aby okamžitě přestali užívat XELODA a kontaktovali svého poskytovatele zdravotní péče [viz NEŽÁDOUCÍ REAKCE ].
Horečka a neutropenie
Informujte pacienty, u kterých se objeví horečka 100,5 °F nebo vyšší nebo jiné známky potenciální infekce, aby kontaktovali svého poskytovatele zdravotní péče [viz NEŽÁDOUCÍ REAKCE ].
Embryo-fetální toxicita
Upozorněte ženy na reprodukční potenciál potenciálního rizika pro plod a na používání účinné antikoncepce během léčby přípravkem XELODA 500 mg a po dobu 6 měsíců po poslední dávce. Poraďte ženám, aby informovaly svého poskytovatele zdravotní péče o známém nebo předpokládaném těhotenství [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ , Použití u konkrétních populací ].
Poraďte mužské pacienty s partnerkami s reprodukčním potenciálem, aby používali účinnou antikoncepci během léčby přípravkem XELODA 500 mg a po dobu 3 měsíců po poslední dávce (viz Použití u konkrétních populací ].
Laktace
Doporučte ženám, aby během léčby přípravkem XELODA 500 mg a 2 týdny po poslední dávce nekojily (viz Použití u konkrétních populací ].
Neklinická toxikologie
Karcinogeneze, mutageneze, zhoršení plodnosti
Nebyly provedeny adekvátní studie zkoumající karcinogenní potenciál kapecitabinu. Kapecitabin nebyl in vitro mutagenní vůči bakteriím (Amesův test) nebo savčím buňkám (test genové mutace čínského křečka V79/HPRT). Kapecitabin byl klastogenní in vitro pro lidské lymfocyty periferní krve, ale ne klastogenní in vivo pro myší kostní dřeň (mikronukleární test). Fluoruracil způsobuje mutace bakterií a kvasinek. Fluoruracil také způsobuje chromozomální abnormality v mikronukleárním testu u myší in vivo.
Ve studiích fertility a obecné reprodukční schopnosti u myších samic narušily perorální dávky kapecitabinu 760 mg/kg/den (asi 2300 mg/m2/den) estrus a následně způsobily pokles fertility. U myší, které otěhotněly, tuto dávku nepřežil žádný plod. Porucha estru byla reverzibilní. U mužů tato dávka způsobila degenerativní změny ve varlatech, včetně snížení počtu spermatocytů a spermatid. V samostatných farmakokinetických studiích tato dávka u myší vyvolala hodnoty AUC 5'-DFUR přibližně 0,7krát vyšší než odpovídající hodnoty u pacientů, kterým byla podávána doporučená denní dávka.
Použití u konkrétních populací
Těhotenství
Shrnutí rizik
Na základě zjištění v reprodukčních studiích na zvířatech a mechanismu účinku může XELODA 500 mg způsobit poškození plodu při podání těhotné ženě (viz KLINICKÁ FARMAKOLOGIE ]. Omezené dostupné údaje u lidí nejsou dostatečné k tomu, aby informovaly o riziku spojeném s užíváním léčiva během těhotenství. V reprodukčních studiích na zvířatech způsobilo podávání kapecitabinu březím zvířatům během období organogeneze embryonální letalitu a teratogenitu u myší a embryonální letalitu u opic při 0,2 a 0,6násobku expozice (AUC) u pacientů užívajících doporučenou dávku [viz Data ]. Informujte těhotné ženy o potenciálním riziku pro plod.
Odhadované základní riziko závažných vrozených vad a potratu pro indikovanou populaci není známo. V obecné populaci USA je odhadované základní riziko závažných vrozených vad a potratu u klinicky uznaných těhotenství 2–4 % a 15–20 %.
Data
Údaje o zvířatech
Perorální podávání kapecitabinu březím myším během období organogeneze v dávce 198 mg/kg/den způsobilo malformace a embryonální letalitu. V samostatných farmakokinetických studiích tato dávka u myší vyvolala hodnoty AUC 5'-DFUR, které byly přibližně 0,2násobkem hodnot AUC u pacientů, kterým byla podávána doporučená denní dávka. Malformace u myší zahrnovaly rozštěp patra, anoftalmii, mikroftalmii, oligodaktylii, polydaktylii, syndaktylii, zakřivený ocas a dilataci mozkových komor. Perorální podávání kapecitabinu březím opicím během období organogeneze v dávce 90 mg/kg/den způsobilo fetální letalitu. Tato dávka vyvolala hodnoty AUC 5'-DFUR, které byly přibližně 0,6násobkem hodnot AUC u pacientů, kterým byla podávána doporučená denní dávka.
Laktace
Shrnutí rizik
Neexistují žádné informace o přítomnosti kapecitabinu v mateřském mléce nebo o jeho účincích na produkci mléka nebo na kojené dítě. Metabolity kapecitabinu byly přítomny v mléce kojících myší [viz Data ]. Vzhledem k potenciálu závažných nežádoucích účinků při expozici kapecitabinu u kojených dětí doporučte ženám, aby během léčby přípravkem XELODA 500 mg a 2 týdny po poslední dávce nekojily.
Data
Kojící myši, kterým byla podána jednorázová perorální dávka kapecitabinu, vylučovaly do mléka významné množství metabolitů kapecitabinu.
Ženy a muži reprodukčního potenciálu
Těhotenský test
Před zahájením léčby přípravkem XELODA se u žen s reprodukčním potenciálem doporučuje provést těhotenský test.
Antikoncepce
Ženy
XELODA 500 mg může způsobit poškození plodu při podání těhotné ženě [viz Těhotenství ]. Informujte ženy s reprodukčním potenciálem, aby během léčby a po dobu 6 měsíců po poslední dávce přípravku XELODA používaly účinnou antikoncepci.
Muži
Na základě nálezů genetické toxicity doporučte mužským pacientům s partnerkami s reprodukčním potenciálem používat účinnou antikoncepci během léčby a po dobu 3 měsíců po poslední dávce XELODA (viz Neklinická toxikologie ].
Neplodnost
Na základě studií na zvířatech může XELODA poškodit plodnost u samic a samců s reprodukčním potenciálem (viz Neklinická toxikologie ].
Pediatrické použití
Bezpečnost a účinnost přípravku XELODA u pediatrických pacientů nebyla stanovena. Ve dvou jednoramenných studiích u pediatrických pacientů s nově diagnostikovanými gliomy mozkového kmene a gliomy vysokého stupně nebyl prokázán žádný klinický přínos. V obou studiích dostávali dětští pacienti zkoumanou pediatrickou formu kapecitabinu současně s radiační terapií a po jejím ukončení (celková dávka 5580 cGy ve frakcích 180 cGy). Relativní biologická dostupnost zkoumané formulace k XELODA 500 mg byla podobná.
První studie byla provedena u 22 dětských pacientů (střední věk 8 let, rozmezí 5–17 let) s nově diagnostikovanými nediseminovanými vnitřními difuzními gliomy mozkového kmene a gliomy vysokého stupně. V části studie zaměřené na zjištění dávky dostávali pacienti kapecitabin souběžně s radiační terapií v dávkách od 500 mg/m2 do 850 mg/m2 každých 12 hodin po dobu až 9 týdnů. Po 2týdenní přestávce pacienti dostávali 1250 mg/m2 kapecitabinu každých 12 hodin ve dnech 1-14 21denního cyklu až po 3 cykly. Maximální tolerovaná dávka (MTD) kapecitabinu podávaného současně s radioterapií byla 650 mg/m2 každých 12 hodin. Hlavními toxicitami limitujícími dávku byly palmárno-plantární erytrodysestezie a zvýšení alaninaminotransferázy (ALT).
Druhá studie byla provedena u 34 dalších pediatrických pacientů s nově diagnostikovanými nediseminovanými vnitřními difuzními gliomy mozkového kmene (střední věk 7 let, rozmezí 3–16 let) au 10 pediatrických pacientů, kteří dostávali MTD kapecitabinu ve studii pro stanovení dávky a splnili kritéria způsobilosti pro tento pokus. Všichni pacienti dostávali kapecitabin v dávce 650 mg/m2 každých 12 hodin se současnou radioterapií po dobu až 9 týdnů. Po 2týdenní přestávce pacienti dostávali 1250 mg/m2 kapecitabinu každých 12 hodin ve dnech 1-14 21denního cyklu až po 3 cykly.
U pediatrických pacientů s nově diagnostikovanými intrinsickými gliomy mozkového kmene, kteří dostávali kapecitabin, nedošlo k žádnému zlepšení v jednoleté míře přežití bez progrese a jednoleté celkové míře přežití ve srovnání s podobnou populací pediatrických pacientů, kteří se účastnili jiných klinických studií.
Profil nežádoucích účinků kapecitabinu byl v souladu se známým profilem nežádoucích účinků u dospělých, s výjimkou laboratorních abnormalit, které se vyskytovaly častěji u pediatrických pacientů. Nejčastěji hlášené laboratorní abnormality (výskyt na pacienta ≥ 40 %) byly zvýšená ALT (75 %), lymfocytopenie (73 %), leukopenie (73 %), hypokalémie (68 %), trombocytopenie (57 %), hypoalbuminémie (55 %), neutropenie (50 %), nízký hematokrit (50 %), hypokalcémie (48 %), hypofosfatemie (45 %) a hyponatremie (45 %).
Geriatrické použití
Lékaři by měli věnovat zvláštní pozornost sledování nežádoucích účinků přípravku XELODA 500 mg u starších osob (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Jaterní nedostatečnost
Pokud jsou XELODA léčeni pacienti s mírnou až středně těžkou jaterní dysfunkcí způsobenou jaterními metastázami, postupujte opatrně. Účinek těžké jaterní dysfunkce na XELODA není znám (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ a KLINICKÁ FARMAKOLOGIE ].
Renální insuficience
Pacienti se středně těžkou (clearance kreatininu = 30 až 50 ml/min) a těžkou (clearance kreatininu KONTRAINDIKACE , VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ , DÁVKOVÁNÍ A PODÁVÁNÍ , a KLINICKÁ FARMAKOLOGIE ].
PŘEDÁVKOVAT
Mezi projevy akutního předávkování patří nevolnost, zvracení, průjem, gastrointestinální podráždění a krvácení a útlum kostní dřeně. Medikamentózní léčba předávkování by měla zahrnovat obvyklé podpůrné lékařské zásahy zaměřené na nápravu přítomných klinických projevů. Ačkoli nebyly hlášeny žádné klinické zkušenosti s použitím dialýzy jako léčby předávkování XELODA 500 mg, dialýza může být přínosem pro snížení cirkulujících koncentrací 5'-DFUR, nízkomolekulárního metabolitu mateřské látky.
Jednotlivé dávky XELODA 500 mg nebyly smrtelné pro myši, potkany a opice v dávkách až 2 000 mg/kg (2,4, 4,8 a 9,6násobek doporučené denní dávky pro člověka na základě mg/m2).
KONTRAINDIKACE
Těžké poškození ledvin
XELODA je kontraindikována u pacientů s těžkou poruchou funkce ledvin (clearance kreatininu pod 30 ml/min [Cockroft a Gault]) (viz Použití u konkrétních populací a KLINICKÁ FARMAKOLOGIE ].
Přecitlivělost
Přípravek XELODA 500 mg je kontraindikován u pacientů se známou přecitlivělostí na kapecitabin nebo na kteroukoli jeho složku. Přípravek XELODA je kontraindikován u pacientů se známou přecitlivělostí na 5-fluorouracil.
KLINICKÁ FARMAKOLOGIE
Mechanismus působení
Enzymy přeměňují kapecitabin na 5-fluorouracil (5-FU) in vivo. Normální i nádorové buňky metabolizují 5-FU na 5-fluor-2'-deoxyuridinmonofosfát (FdUMP) a 5-fluoruridintrifosfát (FUTP). Tyto metabolity způsobují poškození buněk dvěma různými mechanismy. Nejprve se FdUMP a folátový kofaktor, N5-10-methylentetrahydrofolát, vážou na thymidylátsyntázu (TS) za vzniku kovalentně vázaného ternárního komplexu. Tato vazba inhibuje tvorbu thymidylátu z 2'-deoxyuridylátu. Thymidylát je nezbytným prekurzorem thymidintrifosfátu, který je nezbytný pro syntézu DNA, takže nedostatek této sloučeniny může inhibovat buněčné dělení. Za druhé, jaderné transkripční enzymy mohou omylem začlenit FUTP místo uridintrifosfátu (UTP) během syntézy RNA. Tato metabolická chyba může interferovat se zpracováním RNA a syntézou proteinů.
Farmakokinetika
Vstřebávání
Po perorálním podání 1255 mg/m2 BID pacientům s rakovinou dosáhl kapecitabin maximálních hladin v krvi přibližně za 1,5 hodiny (Tmax), přičemž maximální hladiny 5-FU se objevily o něco později, za 2 hodiny. Jídlo snižovalo jak rychlost, tak rozsah absorpce kapecitabinu, přičemž průměrná Cmax a AUC0-∞ se snížily o 60 % a 35 %. Cmax a AUC0-∞ 5-FU byly také sníženy jídlem o 43 %, resp. 21 %. Jídlo zpozdilo Tmax jak rodiče, tak 5-FU o 1,5 hodiny [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ , DÁVKOVÁNÍ A PODÁVÁNÍ , a Interakce droga-jídlo ].
Farmakokinetika XELODA 500 mg a jejích metabolitů byla hodnocena u přibližně 200 pacientů s rakovinou v rozmezí dávek 500 až 3500 mg/m2/den. V tomto rozmezí byla farmakokinetika XELODA a jejího metabolitu 5'-DFCR úměrná dávce a v průběhu času se neměnila. Zvýšení AUC 5'-DFUR a 5-FU však bylo větší než úměrné zvýšení dávky a AUC 5-FU byla 14. den o 34 % vyšší než 1. den. Cmax a AUC 5-FU byly vyšší než 85 %.
Rozdělení
Vazba kapecitabinu a jeho metabolitů na plazmatické proteiny je nižší než 60 % a není závislá na koncentraci. Kapecitabin se primárně vázal na lidský albumin (přibližně 35 %). XELODA 500 mg má nízký potenciál pro farmakokinetické interakce související s vazbou na plazmatické proteiny.
Bioaktivace a metabolismus
Kapecitabin je rozsáhle metabolizován enzymaticky na 5-FU. V játrech 60 kDa karboxylesteráza hydrolyzuje velkou část sloučeniny na 5'-deoxy-5-fluorocytidin (5'-DFCR). Cytidindeamináza, enzym nacházející se ve většině tkání, včetně nádorů, následně přeměňuje 5'-DFCR na 5'-DFUR. Enzym, thymidin fosforyláza (dThdPase), pak hydrolyzuje 5'DFUR na aktivní léčivo 5-FU. Mnoho tkání v celém těle exprimuje thymidinfosforylázu. Některé lidské karcinomy exprimují tento enzym ve vyšších koncentracích než okolní normální tkáně. Po perorálním podání XELODA 7 dní před chirurgickým zákrokem u pacientů s kolorektálním karcinomem byl střední poměr koncentrace 5-FU v kolorektálních nádorech k přilehlým tkáním 2,9 (rozsah od 0,9 do 8,0). Tyto poměry nebyly hodnoceny u pacientů s rakovinou prsu ani nebyly srovnávány s infuzí 5-FU.
Metabolická cesta kapecitabinu na 5-FU
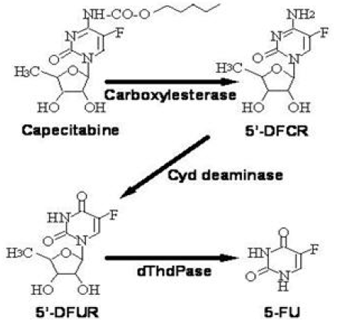
Enzym dihydropyrimidindehydrogenáza hydrogenuje 5-FU, produkt metabolismu kapecitabinu, na mnohem méně toxický 5-fluor-5,6-dihydro-fluoruracil (FUH2). Dihydropyrimidináza štěpí pyrimidinový kruh za vzniku kyseliny 5-fluor-ureido-propionové (FUPA). Nakonec β-ureido-propionáza štěpí FUPA na α-fluor-β-alanin (FBAL), který je odstraněn močí.
In vitro enzymatické studie s mikrozomy lidských jater ukázaly, že kapecitabin a jeho metabolity (5'-DFUR, 5'-DFCR, 5-FU a FBAL) neinhibují metabolismus testovaných substrátů pomocí izoenzymů 1A2, 2A6, 3A4 cytochromu P450, 2C19, 2D6 a 2E1.
Vylučování
Kapecitabin a jeho metabolity se vylučují převážně močí; 95,5 % podané dávky kapecitabinu se objeví v moči. Vylučování stolicí je minimální (2,6 %). Hlavním metabolitem vylučovaným močí je FBAL, který představuje 57 % podané dávky. Asi 3 % podané dávky se vyloučí močí jako nezměněné léčivo. Eliminační poločas původního kapecitabinu i 5-FU byl asi 0,75 hodiny.
Vliv věku, pohlaví a rasy na farmakokinetiku kapecitabinu
Populační analýza shromážděných údajů ze dvou velkých kontrolovaných studií u pacientů s metastatickým kolorektálním karcinomem (n=505), kterým byla podávána XELODA 500 mg v dávce 1250 mg/m2 dvakrát denně, ukázala, že pohlaví (202 žen a 303 mužů) a rasa (455 bílých/bělošských pacientů, 22 černých pacientů a 28 pacientů jiné rasy) nemají žádný vliv na farmakokinetiku 5'DFUR, 5-FU a FBAL. Věk nemá významný vliv na farmakokinetiku 5'-DFUR a 5-FU v rozmezí 27 až 86 let. 20% zvýšení věku má za následek 15% zvýšení AUC FBAL [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ a DÁVKOVÁNÍ A PODÁVÁNÍ ].
Po perorálním podávání kapecitabinu v dávce 825 mg/m2 dvakrát denně po dobu 14 dnů měli japonští pacienti (n=18) asi o 36 % nižší Cmax a o 24 % nižší AUC kapecitabinu než bělošští pacienti (n=22). Japonští pacienti měli také asi o 25 % nižší Cmax a o 34 % nižší AUC pro FBAL než bělošští pacienti. Klinický význam těchto rozdílů není znám. V expozici jiným metabolitům (5'-DFCR, 5'-DFUR a 5FU) nedošlo k žádným významným rozdílům.
Účinek jaterní nedostatečnosti
XELODA byla hodnocena u 13 pacientů s mírnou až středně těžkou jaterní dysfunkcí způsobenou jaterními metastázami definovaným složeným skóre zahrnujícím bilirubin, AST/ALT a alkalickou fosfatázu po jednorázové dávce 1255 mg/m2 XELODA. AUC0-∞ i Cmax kapecitabinu se u pacientů s jaterní dysfunkcí zvýšily o 60 % ve srovnání s pacienty s normální funkcí jater (n=14). AUC0-∞ a Cmax 5-FU nebyly ovlivněny. U pacientů s mírnou až středně závažnou jaterní dysfunkcí způsobenou jaterními metastázami je třeba při podávání XELODA 500 mg postupovat opatrně. Účinek těžké jaterní dysfunkce na XELODA 500 mg není znám (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ a Použití ve zvláštních populacích ].
Vliv Renální Insuficience
Po perorálním podávání kapecitabinu v dávce 1 250 mg/m2 dvakrát denně pacientům s rakovinou s různým stupněm poruchy funkce ledvin se u pacientů se středně těžkou (clearance kreatininu = 30 až 50 ml/min) a těžkou (clearance kreatininu 80 ml/min). Systémová expozice 5'-DFUR byla o 42 % a 71 % vyšší u pacientů se středně těžkou a těžkou poruchou funkce ledvin než u normálních pacientů. Systémová expozice kapecitabinu byla asi o 25 % vyšší u pacientů se středně těžkou i těžkou poruchou funkce ledvin (viz DÁVKOVÁNÍ A PODÁVÁNÍ , KONTRAINDIKACE , VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ , a Použití ve zvláštních populacích ].
Vliv kapecitabinu na farmakokinetiku warfarinu
čtyř pacientů s rakovinou zvýšilo chronické podávání kapecitabinu (1250 mg/m2 dvakrát denně) s jednorázovou dávkou 20 mg warfarinu průměrnou AUC S-warfarinu o 57 % a snížilo jeho clearance o 37 %. Výchozí korigovaná AUC INR se u těchto 4 pacientů zvýšila 2,8krát a maximální pozorovaná střední hodnota INR se zvýšila o 91 % [viz BOX VAROVÁNÍ a DROGOVÉ INTERAKCE ].
Vliv antacidů na farmakokinetiku kapecitabinu
Když byl Maalox® (20 ml), antacidum obsahující hydroxid hlinitý a hydroxid hořečnatý, podán bezprostředně po XELODA (1250 mg/m2, n=12 pacientů s rakovinou), AUC a Cmax se zvýšily o 16 % a 35 %, v tomto pořadí, u kapecitabinu ao 18 % a 22 % u 5'-DFCR. Na ostatní tři hlavní metabolity (5'-DFUR, 5-FU, FBAL) XELODA nebyl pozorován žádný účinek.
Účinek alopurinolu na kapecitabin
Publikovaná literatura uvádí, že současné užívání s alopurinolem může snížit konverzi kapecitabinu na aktivní metabolity FdUMP a FUTP; klinický význam však nebyl plně charakterizován.
Vliv kapecitabinu na farmakokinetiku docetaxelu a naopak
Studie fáze 1 hodnotila účinek XELODA na farmakokinetiku docetaxelu (Taxotere®) a účinek docetaxelu na farmakokinetiku XELODA byla provedena u 26 pacientů se solidními nádory. Bylo zjištěno, že XELODA nemá žádný vliv na farmakokinetiku docetaxelu (Cmax a AUC) a docetaxel nemá žádný vliv na farmakokinetiku kapecitabinu a 5-FU prekurzoru 5'-DFUR.
Klinické studie
Adjuvantní rakovina tlustého střeva
Multicentrická randomizovaná, kontrolovaná klinická studie fáze 3 u pacientů s Dukesovým C karcinomem tlustého střeva (X-ACT) poskytla údaje týkající se použití přípravku XELODA k adjuvantní léčbě pacientů s karcinomem tlustého střeva. Primárním cílem studie bylo porovnat přežití bez onemocnění (DFS) u pacientů užívajících XELODA s těmi, kteří dostávali IV 5-FU/LV samotnou. V této studii bylo 1987 pacientů randomizováno buď k léčbě přípravkem XELODA 1250 mg/m2 perorálně dvakrát denně po dobu 2 týdnů, po nichž následovala 1 týdenní přestávka, podávaná ve 3týdenních cyklech celkem 8 cyklů (24 týdnů) nebo IV. bolus 5-FU 425 mg/m2 a 20 mg/m2 IV leukovorinu ve dnech 1 až 5, podávaných ve 4týdenních cyklech celkem na 6 cyklů (24 týdnů). Pacienti ve studii museli být ve věku 18 až 75 let s histologicky potvrzeným Dukesovým karcinomem tlustého střeva stadia C s alespoň jednou pozitivní lymfatickou uzlinou a museli podstoupit (do 8 týdnů před randomizací) kompletní resekci primárního nádoru bez makroskopického nebo mikroskopického důkazu zbývajícího nádoru. Pacienti také nemuseli podstoupit žádnou předchozí cytotoxickou chemoterapii nebo imunoterapii (kromě steroidů) a měli výkonnostní stav ECOG 0 nebo 1 (KPS ≥ 70 %), ANC ≥ 1,5x109/l, krevní destičky ≥ 100x109/l, sérový kreatinin ≤ 1,5 ULN, celkový bilirubin ≤ 1,5 ULN, AST/ALT ≤ 2,5 ULN a CEA v normálních mezích v době randomizace.
Základní demografické údaje pro pacienty s XELODA a 5-FU/LV jsou uvedeny v tabulce 10. Základní charakteristiky byly mezi rameny dobře vyvážené.
Všichni pacienti s normální funkcí ledvin nebo mírnou poruchou funkce ledvin zahájili léčbu plnou počáteční dávkou 1250 mg/m2 perorálně dvakrát denně. Počáteční dávka byla snížena u pacientů se středně těžkou poruchou funkce ledvin (vypočtená clearance kreatininu 30 až 50 ml/min) na začátku (viz DÁVKOVÁNÍ A PODÁVÁNÍ ]. Následně byly pro všechny pacienty dávky upraveny v případě potřeby podle toxicity. Řízení dávky přípravku XELODA 500 mg zahrnovalo snížení dávky, zpoždění cyklu a přerušení léčby (viz tabulka 11).
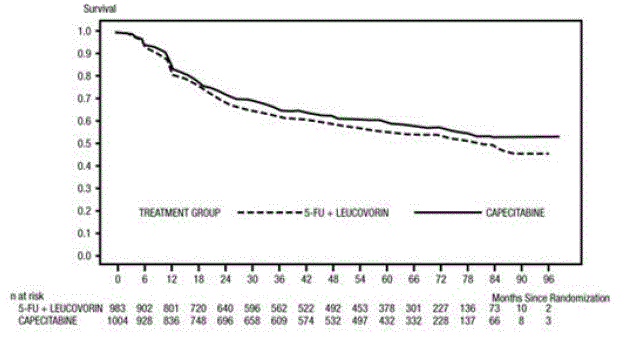
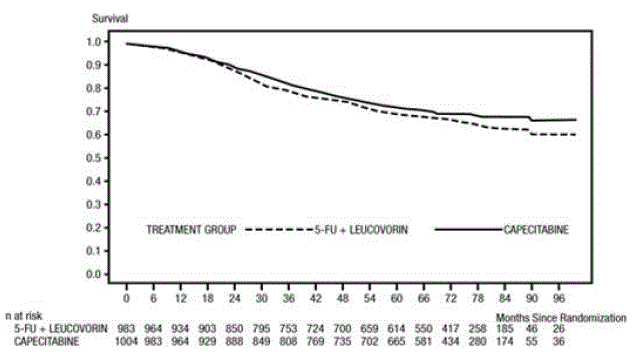
Medián sledování v době analýzy byl 83 měsíců (6,9 roku). Poměr rizik pro DFS pro XELODA 500 mg ve srovnání s 5-FU/LV byl 0,88 (95% CI 0,77 – 1,01) (viz a Obrázek 1 ). Protože horní 2stranná 95% mez spolehlivosti poměru rizika byla menší než 1,20, XELODA nebyla horší než 5-FU/LV. Volba hranice neinferiority 1,20 odpovídá zachování přibližně 75 % účinku 5-FU/LV na DFS. Poměr rizik pro XELODA ve srovnání s 5-FU/LV s ohledem na celkové přežití byl 0,86 (95% CI 0,74 – 1,01). Celkové 5leté přežití bylo 71,4 % pro XELODA a 68,4 % pro 5-FU/LV (viz obrázek 2).
Obrázek 1 Kaplan-Meierovy odhady přežití bez onemocnění (všechny randomizované populace)a
Obrázek 2 Kaplan-Meierovy odhady celkového přežití (veškerá randomizovaná populace)
Metastatický kolorektální karcinom
Všeobecné
Doporučená dávka XELODA 500 mg byla stanovena v otevřené, randomizované klinické studii zkoumající účinnost a bezpečnost kontinuální léčby kapecitabinem (1331 mg/m2/den ve dvou rozdělených dávkách, n=39), intermitentní léčby kapecitabinem ( 2510 mg/m2/den ve dvou rozdělených dávkách, n=34) a intermitentní léčba kapecitabinem v kombinaci s perorálním leukovorinem (LV) (kapecitabin 1657 mg/m2/den ve dvou rozdělených dávkách, n=35; leukovorin 60 mg/ den) u pacientů s pokročilým a/nebo metastatickým kolorektálním karcinomem v první linii metastáz. Nebyla zjištěna žádná zjevná výhoda v míře odpovědi na přidání leukovorinu k přípravku XELODA; toxicita se však zvýšila. XELODA, 1250 mg/m2 dvakrát denně po dobu 14 dnů s následnou 1 týdenní přestávkou, byla vybrána pro další klinický vývoj na základě celkového profilu bezpečnosti a účinnosti tří studovaných schémat.
Monoterapie
Údaje ze dvou otevřených, multicentrických, randomizovaných, kontrolovaných klinických studií zahrnujících 1207 pacientů podporují použití přípravku XELODA v první linii léčby pacientů s metastatickým kolorektálním karcinomem. Tyto dvě klinické studie měly identický design a byly provedeny ve 120 centrech v různých zemích. Studie 1 byla provedena v USA, Kanadě, Mexiku a Brazílii; Studie 2 byla provedena v Evropě, Izraeli, Austrálii, Novém Zélandu a na Tchaj-wanu. Celkem bylo v obou studiích randomizováno 603 pacientů k léčbě přípravkem XELODA 500 mg v dávce 1250 mg/m2 dvakrát denně po dobu 2 týdnů, po nichž následovala 1 týdenní přestávka a podávaná ve 3týdenních cyklech; 604 pacientů bylo randomizováno k léčbě 5-FU a leukovorinem (20 mg/m2 leukovorinu IV a následně 425 mg/m2 IV bolus 5-FU, ve dnech 1 až 5, každých 28 dní).
obou studiích bylo hodnoceno celkové přežití, doba do progrese a míra odpovědi (kompletní plus částečné odpovědi). Odpovědi byly definovány kritérii Světové zdravotnické organizace a předloženy zaslepené nezávislé hodnotící komisi (IRC). Rozdíly v hodnocení mezi zkoušejícím a IRC byly srovnány sponzorem, zaslepeným vůči léčebné větvi, podle specifikovaného algoritmu. Přežití bylo hodnoceno na základě analýzy non-inferiority.
Základní demografické údaje pro pacienty s XELODA a 5-FU/LV jsou uvedeny v tabulce 13.
Koncové body účinnosti pro dvě studie fáze 3 jsou uvedeny v a
Obrázek 3 Kaplan-Meierova křivka pro celkové přežití sdružených dat (studie 1 a 2)
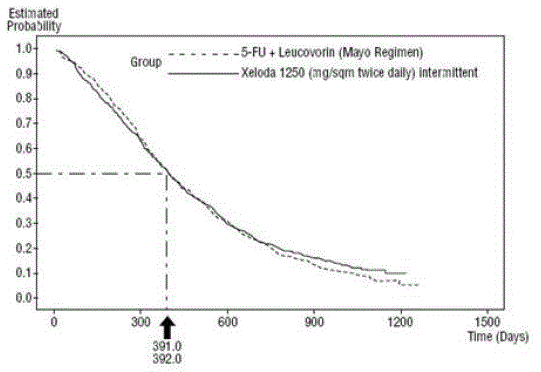
XELODA byla lepší než 5-FU/LV v míře objektivní odpovědi ve studii 1 a studii 2. Podobnost XELODA 500 mg a 5-FU/LV v těchto studiích byla hodnocena zkoumáním potenciálního rozdílu mezi těmito dvěma způsoby léčby. Aby bylo zajištěno, že přípravek XELODA 500 mg má klinicky významný účinek na přežití, byly provedeny statistické analýzy ke stanovení procenta účinku 5-FU/LV na přežití, který si přípravek XELODA zachoval. Odhad účinku 5-FU/LV na přežití byl odvozen z metaanalýzy deseti randomizovaných studií z publikované literatury porovnávající 5-FU s režimy 5-FU/LV, které byly podobné kontrolním ramenům použitým v těchto studiích 1 a 2. Metodou pro srovnání léčby bylo prozkoumat nejhorší případ (95% horní hranice spolehlivosti) pro rozdíl mezi 5-FU/LV a XELODA 500 mg a ukázat, že ztráta více než 50 % z 5- Účinek přežití FU/LV byl vyloučen. Bylo prokázáno, že procento udržovaného účinku 5-FU/LV na přežití bylo alespoň 61 % pro studii 2 a 10 % pro studii 1. Souhrnný výsledek je v souladu se zachováním alespoň 50 % účinku 5 -FU/LV. Je třeba poznamenat, že tyto hodnoty pro zachovaný účinek jsou založeny na horní hranici rozdílu 5-FU/LV oproti XELODA 500 mg. Tyto výsledky nevylučují možnost skutečné ekvivalence přípravku XELODA 500 mg a 5-FU/LV (viz a Obrázek 3 ).
Rakovina prsu
XELODA byla hodnocena v klinických studiích v kombinaci s docetaxelem (Taxotere®) a jako monoterapie.
V kombinaci s docetaxelem
Dávka XELODA použitá v klinické studii fáze 3 v kombinaci s docetaxelem vycházela z výsledků studie fáze 1, kde byl rozsah dávek docetaxelu podávaných ve 3týdenních cyklech v kombinaci s intermitentním režimem XELODA (14 dní léčby, po které následovala 7denní přestávka). Kombinovaný dávkový režim byl zvolen na základě profilu snášenlivosti 75 mg/m2 podávaného ve 3týdenních cyklech docetaxelu v kombinaci s 1250 mg/m2 dvakrát denně po dobu 14 dnů XELODA 500 mg podávanou ve 3týdenních cyklech. Schválená dávka 100 mg/m2 docetaxelu podávaná ve 3týdenních cyklech byla kontrolní větví 3. fáze studie.
XELODA v kombinaci s docetaxelem byla hodnocena v otevřené, multicentrické, randomizované studii v 75 centrech v Evropě, Severní Americe, Jižní Americe, Asii a Austrálii. Celkem bylo zařazeno 511 pacientek s metastazujícím karcinomem prsu rezistentním nebo recidivujícím během nebo po léčbě obsahující antracykliny nebo s relapsem během nebo recidivující během 2 let po dokončení adjuvantní léčby obsahující antracykliny. Dvě stě padesát pět (255) pacientů bylo randomizováno k podávání XELODA 1250 mg/m2 dvakrát denně po dobu 14 dnů, po nichž následoval 1 týden bez léčby a docetaxelu 75 mg/m2 ve formě 1hodinové intravenózní infuze podávané ve 3týdenních cyklech. V rameni s monoterapií dostávalo 256 pacientů docetaxel 100 mg/m2 ve formě 1hodinové intravenózní infuze podávané ve 3týdenních cyklech. Demografické údaje pacientů jsou uvedeny v tabulce 16.
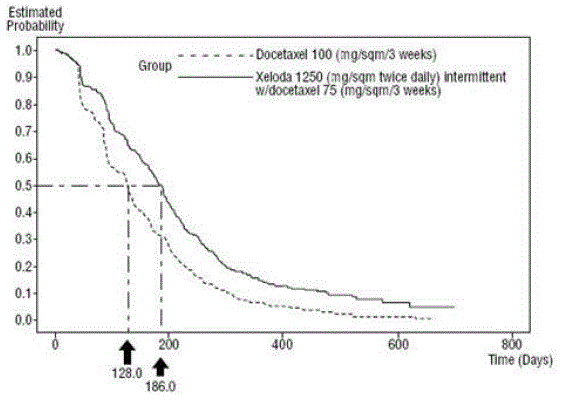
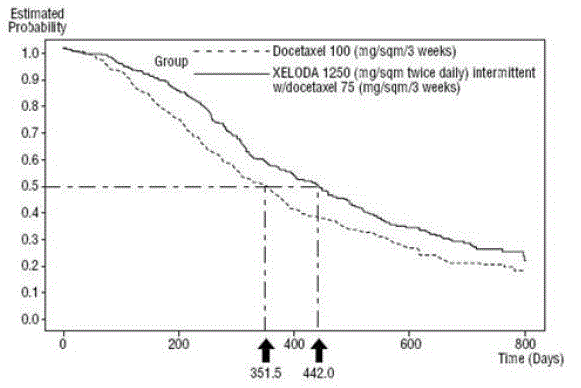
XELODA v kombinaci s docetaxelem vedla ke statisticky významnému zlepšení doby do progrese onemocnění, celkového přežití a míry objektivní odpovědi ve srovnání s monoterapií docetaxelem, jak je uvedeno v a Obrázek 5 .
Obrázek 4 Kaplan-Meierovy odhady doby do progrese onemocnění XELODA a docetaxel vs. docetaxel
Obrázek 5 Kaplan-Meierovy odhady přežití XELODA a docetaxel vs. docetaxel
Monoterapie
Protinádorová aktivita XELODA jako monoterapie byla hodnocena v otevřené jednoramenné studii provedené ve 24 centrech v USA a Kanadě. Celkem bylo zařazeno 162 pacientek s karcinomem prsu stadia IV. Primárním cílovým parametrem byla míra odpovědi nádoru u pacientů s měřitelným onemocněním, přičemž odpověď byla definována jako ≥50% snížení součtu součinů kolmých průměrů dvourozměrně měřitelného onemocnění po dobu alespoň 1 měsíce. XELODA byla podávána v dávce 1255 mg/m2 dvakrát denně po dobu 2 týdnů, po nichž následovala 1 týdenní přestávka a podávána ve 3týdenních cyklech. Základní demografické údaje a klinické charakteristiky všech pacientů (n=162) a pacientů s měřitelným onemocněním (n=135) jsou uvedeny v . Rezistence byla definována jako progresivní onemocnění během léčby, s počáteční odpovědí nebo bez ní, nebo jako relaps během 6 měsíců po dokončení léčby adjuvantním chemoterapeutickým režimem obsahujícím antracykliny.
Protinádorové odpovědi u pacientů s onemocněním rezistentním na paclitaxel i antracyklin jsou uvedeny v
U podskupiny 43 pacientů, kteří byli dvojnásobně rezistentní, byl medián doby do progrese 102 dnů a medián přežití 255 dnů. Míra objektivní odpovědi v této populaci byla podpořena mírou odpovědi 18,5 % (1 ČR, 24 PR) v celkové populaci 135 pacientů s měřitelným onemocněním, kteří byli méně odolní vůči chemoterapii (viz ). Medián doby do progrese byl 90 dnů a medián přežití byl 306 dnů.
INFORMACE PRO PACIENTA
XELODA® (zeh-LOE-duh) (kapecitabin) tablety
Jaké jsou nejdůležitější informace, které bych měl vědět o přípravku XELODA 500 mg?
XELODA 500 mg může způsobit závažné nežádoucí účinky, včetně:
- XELODA může interagovat s léky na ředění krve, jako je warfarin (COUMADIN®). Užívání přípravku XELODA 500 mg s těmito léky může způsobit změny v rychlosti srážení krve a může způsobit krvácení, které může vést ke smrti. K tomu může dojít hned několik dní poté, co začnete užívat přípravek XELODA 500 mg, nebo později během léčby a možná i do 1 měsíce po ukončení užívání přípravku XELODA. Vaše riziko může být vyšší, protože máte rakovinu a pokud je vám více než 60 let.
- Před užitím přípravku XELODA informujte svého poskytovatele zdravotní péče, pokud užíváte warfarin (COUMADIN) nebo jiný lék na ředění krve.
- Pokud během léčby přípravkem XELODA užíváte warfarin (COUMADIN) nebo jiný lék na ředění krve, který je podobný warfarinu (COUMADIN), váš poskytovatel zdravotní péče by měl často provádět krevní testy, aby zkontroloval, jak rychle se vaše krev sráží během a po ukončení léčby přípravkem XELODA. Váš poskytovatel zdravotní péče může v případě potřeby změnit vaši dávku léku na ředění krve.
Vidět Jaké jsou možné vedlejší účinky přípravku XELODA 500 mg? pro více informací o vedlejších účincích.
Co je XELODA?
XELODA je lék na předpis používaný k léčbě lidí s:
- rakovina tlustého střeva, která se rozšířila do lymfatických uzlin v oblasti blízko tlustého střeva (stadium Dukesova C), po operaci.
- rakovina tlustého střeva nebo konečníku (kolorektální), která se rozšířila do jiných částí těla (metastatická), jako první léčbu vaší rakoviny v této fázi.
- rakovina prsu, která se rozšířila do jiných částí těla (metastazující) spolu s jiným lékem zvaným docetaxel poté, co léčba některými jinými protinádorovými léky nezabrala.
- rakovina prsu, která se rozšířila do jiných částí těla a nezlepšila se po léčbě paklitaxelem a některými dalšími protirakovinnými léky, nebo která již nemůže být léčena některými protirakovinnými léky.
Není známo, zda je přípravek XELODA 500 mg bezpečný a účinný u dětí.
Neužívejte přípravek XELODA, jestliže:
- máte závažné problémy s ledvinami
- jste alergický(á) na kapecitabin, 5-fluorouracil nebo na kteroukoli jeho složku přípravku XELODA. Úplný seznam složek přípravku XELODA naleznete na konci této příbalové informace.
Pokud si nejste jisti, zda máte některý z výše uvedených stavů, poraďte se se svým poskytovatelem zdravotní péče, než začnete přípravek XELODA užívat.
Před užitím přípravku XELODA informujte svého poskytovatele zdravotní péče o všech svých zdravotních potížích, včetně toho, zda:
Vidět "Jaké jsou nejdůležitější informace, které bych měl vědět o XELODA?"
- měli problémy se srdcem
- máte problémy s ledvinami nebo játry
- bylo vám řečeno, že vám chybí enzym DPD (dihydropyrimidindehydrogenáza).
- jste těhotná nebo plánujete otěhotnět. XELODA 500 mg může poškodit vaše nenarozené dítě. Váš poskytovatel zdravotní péče by měl před zahájením léčby přípravkem XELODA provést těhotenský test. Okamžitě informujte svého poskytovatele zdravotní péče, pokud během léčby přípravkem XELODA otěhotníte nebo si myslíte, že byste mohla být těhotná.
- Ženy které jsou schopny otěhotnět, by měly během léčby a 6 měsíců po poslední dávce používat účinnou antikoncepci. Promluvte si se svým poskytovatelem zdravotní péče o možnostech antikoncepce, které by pro vás mohly být během léčby přípravkem XELODA vhodné.
- Muži které mají partnerky, které mohou otěhotnět, by měly během léčby a 3 měsíce po poslední dávce používat účinnou antikoncepci.
- kojíte nebo plánujete kojit. Není známo, zda XELODA přechází do vašeho mateřského mléka. Během léčby přípravkem XELODA 500 mg a 2 týdny po poslední dávce nekojte.
Informujte svého poskytovatele zdravotní péče o všech lécích, které užíváte, včetně léků na předpis a volně prodejných léků, vitamínů a bylinných doplňků. XELODA může ovlivnit způsob účinku jiných léků a jiné léky mohou ovlivnit způsob účinku XELODA.
Znát léky, které užíváte. Uchovávejte si jejich seznam, abyste je mohli ukázat svému poskytovateli zdravotní péče a lékárníkovi, když dostanete nový lék.
Jak mám užívat XELODA 500 mg?
- Užívejte XELODU přesně tak, jak vám řekl váš poskytovatel zdravotní péče.
- Váš poskytovatel zdravotní péče vám řekne, kolik XELODA máte užívat a kdy ji máte užívat.
- Užívejte XELODU 2krát denně, 1krát ráno a 1krát večer.
- Užijte XELODA 500 mg do 30 minut po jídle.
- Tablety XELODA 500 mg polykejte celé a zapijte vodou. Ne rozdrtit nebo nakrájet tablety XELODA 500 mg. Pokud nemůžete tablety XELODA spolknout celé, informujte o tom svého lékaře.
- Pokud se u Vás objeví nežádoucí účinky, Váš poskytovatel zdravotní péče může změnit Vaši dávku, dočasně přerušit nebo trvale ukončit léčbu přípravkem XELODA 500 mg.
- Pokud užijete příliš mnoho přípravku XELODA, zavolejte svého poskytovatele zdravotní péče nebo jděte ihned na pohotovost v nejbližší nemocnici.
Jaké jsou možné vedlejší účinky přípravku XELODA?
XELODA může způsobit závažné nežádoucí účinky včetně:
Nevolnost a zvracení jsou u přípravku XELODA běžné. Pokud ztratíte chuť k jídlu, cítíte se slabí a máte nevolnost, zvracení nebo průjem, můžete se rychle dehydratovat.
Přestaňte užívat přípravek XELODA a ihned kontaktujte svého poskytovatele zdravotní péče, pokud:
Pokud je počet vašich bílých krvinek velmi nízký, máte zvýšené riziko infekce. Okamžitě zavolejte svého poskytovatele zdravotní péče, pokud se u vás objeví horečka 100,5 °F nebo vyšší nebo máte jiné známky a příznaky infekce.
- Vidět "Jaké jsou nejdůležitější informace, které bych měl vědět o XELODA?"
- Průjem. Průjem je u XELODA běžný a může být někdy závažný. Přestaňte užívat přípravek XELODA a ihned kontaktujte svého poskytovatele zdravotní péče, pokud se počet stolic, které máte za den, zvýší o 4 nebo více stolic než je obvyklé pro vás nebo stolice v noci. Zeptejte se svého poskytovatele zdravotní péče na to, jaké léky můžete užívat k léčbě průjmu. Pokud máte těžký krvavý průjem se silnou bolestí břicha a horečkou, přestaňte Xeloda 500 mg užívat a zavolejte svému poskytovateli zdravotní péče nebo jděte na pohotovost v nejbližší nemocnici.
- Srdeční problémy. XELODA může způsobit srdeční problémy včetně: srdečního záchvatu a sníženého průtoku krve srdcem, bolesti na hrudi, nepravidelného srdečního tepu, změn v elektrické aktivitě vašeho srdce patrných na elektrokardiogramu (EKG), problémů se srdečním svalem, srdečního selhání a náhlého smrt. Přestaňte užívat přípravek XELODA 500 mg a zavolejte svého poskytovatele zdravotní péče nebo jděte ihned na pohotovost v nejbližší nemocnici, pokud se u Vás objeví jakékoli nové příznaky srdečních potíží, včetně:
- bolest na hrudi
- závrať
- dušnost
- točení hlavy
- Ztráta příliš velkého množství tělesných tekutin (dehydratace) a selhání ledvin. Při užívání přípravku XELODA 500 mg může dojít k dehydrataci a může způsobit náhlé selhání ledvin, které může vést ke smrti. Máte vyšší riziko, pokud máte problémy s ledvinami před užitím přípravku XELODA 500 mg a také užíváte jiné léky, které mohou způsobit problémy s ledvinami.
- zvracet 2 nebo vícekrát za den.
- mohou jíst nebo pít jen občas, nebo vůbec kvůli nevolnosti.
- mít průjem. Viz „průjem“ výše.
- Závažné reakce kůže a úst.
- XELODA 500 mg může způsobit závažné kožní reakce, které mohou vést ke smrti. Okamžitě informujte svého poskytovatele zdravotní péče, pokud se u vás objeví kožní vyrážka, puchýře a olupování kůže. Váš poskytovatel zdravotní péče vám může sdělit, abyste přestal(a) užívat přípravek XELODA, pokud máte závažnou kožní reakci. Pokud k tomu dojde, neužívejte přípravek XELODA 500 mg znovu.
- XELODA 500 mg může také způsobit syndrom „ruky a nohy“. Syndrom rukou a nohou je u přípravku XELODA 500 mg běžný a může způsobit znecitlivění a změny citlivosti rukou a nohou nebo způsobit zarudnutí, bolest, otoky rukou a nohou. Přestaňte užívat přípravek XELODA a ihned zavolejte svého poskytovatele zdravotní péče, pokud máte některý z těchto příznaků a nejste schopen vykonávat své obvyklé činnosti.
- Syndrom rukou a nohou může vést ke ztrátě otisků prstů, což by mohlo ovlivnit vaši identifikaci.
- můžete mít vřídky v ústech nebo na jazyku při užívání přípravku XELODA. Přestaňte užívat XELODA 500 mg a zavolejte svého poskytovatele zdravotní péče, pokud se u Vás objeví bolestivé zarudnutí, otok nebo vředy v ústech nebo na jazyku, nebo pokud máte problémy s jídlem.
- Zvýšená hladina bilirubinu v krvi a problémy s játry. Zvýšení bilirubinu v krvi je běžné u přípravku XELODA 500 mg a může být také někdy závažné. Váš poskytovatel zdravotní péče vás během léčby přípravkem XELODA zkontroluje, zda nemáte tyto problémy.
- Snížený počet bílých krvinek, krevních destiček a červených krvinek. Váš poskytovatel zdravotní péče vám během léčby přípravkem XELODA provede krevní testy, aby zkontroloval počet vašich krvinek.
U lidí ve věku 80 let nebo starších může být pravděpodobnější, že se u přípravku XELODA vyvinou závažné nebo závažné nežádoucí účinky.
Mezi nejčastější nežádoucí účinky přípravku XELODA 500 mg patří:
- průjem
- bolest v oblasti žaludku (břicha).
- syndrom ruky a nohy
- slabost a únava
- nevolnost
- zvracel
- zvýšené množství produktů rozpadu červených krvinek (bilirubinu v krvi
Při užívání přípravku XELODA se mohou vyskytnout závažné alergické reakce. Informujte svého poskytovatele zdravotní péče, pokud jste někdy měli alergickou reakci na kapecitabin nebo 5-fluorouracil. Vidět „Neužívejte XELODU, jestliže:“. Přestaňte užívat přípravek XELODA 500 mg a ihned zavolejte svého poskytovatele zdravotní péče nebo jděte na pohotovost, pokud máte některý z následujících příznaků závažné alergické reakce:
- červené svědivé šrámy na kůži (kopřivka)
- zarudnutí kůže
- otok obličeje, rtů, jazyka nebo hrdla
- vyrážka
- svědění
- potíže s polykáním nebo dýcháním
XELODA 500 mg může způsobit problémy s plodností u žen a mužů. To může ovlivnit schopnost mít dítě. Promluvte si se svým poskytovatelem zdravotní péče, pokud máte obavy o plodnost.
Toto nejsou všechny možné vedlejší účinky přípravku XELODA.
Zavolejte svého lékaře o radu ohledně nežádoucích účinků. Nežádoucí účinky můžete hlásit úřadu FDA na čísle 1-800-FDA-1088.
Jak mám XELODA uchovávat?
- Uchovávejte XELODA 500 mg při pokojové teplotě mezi 68 °F až 77 °F (20 °C až 25 °C).
- Uchovávejte XELODA 500 mg v těsně uzavřené nádobě.
- Zeptejte se svého poskytovatele zdravotní péče nebo lékárníka, jak bezpečně zlikvidovat nepoužitý přípravek XELODA.
Uchovávejte XELODA a všechny léky mimo dosah dětí.
Obecné informace o bezpečném a efektivním používání XELODA.
Léky jsou někdy předepisovány pro jiné účely, než které jsou uvedeny v příbalové informaci pro pacienty. Nepoužívejte XELODA pro stav, pro který nebyl předepsán. Nedávejte XELODA 500 mg jiným lidem, i když mají stejné příznaky jako vy. Může jim to ublížit. Můžete požádat svého lékárníka nebo poskytovatele zdravotní péče o informace o přípravku XELODA 500 mg, který je určen pro zdravotníky.
Jaké jsou složky přípravku XELODA 500 mg?
Aktivní složka: kapecitabin
Neaktivní složky: bezvodá laktóza, sodná sůl kroskarmelózy, hydroxypropylmethylcelulóza, mikrokrystalická celulóza, magnesium-stearát a čištěná voda. Broskvový nebo světle broskvový filmový povlak obsahuje hydroxypropylmethylcelulózu, mastek, oxid titaničitý a syntetické žluté a červené oxidy železa.
Další informace najdete na http://www.gene.com/patients/medicines/xeloda 500mg nebo volejte 1-877-436-3683.
Tyto informace pro pacienty byly schváleny americkým Úřadem pro kontrolu potravin a léčiv.