Epivir 150mg Lamivudine Použití, vedlejší účinky a dávkování. Cena v internetové lékárně. Generické léky bez předpisu.
Co je Epivir a jak se používá?
Epivir 150 mg je lék na předpis používaný k léčbě příznaků infekce HIV a chronické hepatitidy B. Epivir 150 mg lze užívat samostatně nebo s jinými léky.
Epivir patří do třídy léků nazývaných činidla proti hepatitidě B/hepatitidě C; HIV, NRTI.
Není známo, zda je přípravek Epivir bezpečný a účinný u dětí mladších 3 měsíců.
Jaké jsou možné nežádoucí účinky přípravku Epivir 150 mg?
Epivir může způsobit závažné nežádoucí účinky včetně:
- kopřivka,
- potíže s dýcháním,
- otok obličeje, rtů, jazyka nebo hrdla,
- neobvyklá bolest svalů,
- bolest břicha,
- zvracení,
- nepravidelná srdeční frekvence,
- závrať,
- cítit zimu,
- slabost,
- únava,
- silná bolest v horní části žaludku šířící se do zad,
- nevolnost,
- rychlý srdeční tep,
- otok kolem střední části,
- pravostranná bolest v horní části žaludku,
- ztráta chuti k jídlu,
- tmavá moč,
- stolice hliněné barvy,
- zežloutnutí kůže nebo očí (žloutenka),
- horečka,
- noční pocení,
- oteklé žlázy,
- opary,
- kašel,
- sípání,
- průjem,
- ztráta váhy,
- potíže s mluvením nebo polykáním,
- problémy s rovnováhou nebo pohybem očí,
- bodavý pocit,
- otok krku nebo hrdla (zvětšená štítná žláza),
- menstruační změny a
- impotence
Pokud máte některý z výše uvedených příznaků, okamžitě vyhledejte lékařskou pomoc.
Mezi nejčastější nežádoucí účinky přípravku Epivir patří:
- nevolnost,
- průjem,
- bolest hlavy,
- horečka,
- únava,
- celkový pocit nemoci,
- bolest ucha nebo plný pocit,
- potíže se sluchem,
- drenáž z ucha,
- nervozita u dítěte,
- ucpaný nos,
- kýchání,
- bolest v krku a
- kašel
Informujte lékaře, pokud máte jakýkoli nežádoucí účinek, který vás obtěžuje nebo který neustupuje.
Toto nejsou všechny možné vedlejší účinky přípravku Epivir. Pro více informací se zeptejte svého lékaře nebo lékárníka.
Zavolejte svého lékaře o radu ohledně nežádoucích účinků. Nežádoucí účinky můžete hlásit úřadu FDA na čísle 1-800-FDA-1088.
VAROVÁNÍ
EXACERBACE HEPATITIDY B a RŮZNÉ PŘÍPRAVKY EPIVIRU.
Exacerbace hepatitidy B
Závažné akutní exacerbace hepatitidy B byly hlášeny u pacientů, kteří byli současně infikováni virem hepatitidy B (HBV) a virem lidské imunodeficience (HIV-1) a kteří přerušili léčbu přípravkem EPIVIR. U pacientů, kteří přeruší léčbu přípravkem EPIVIR a jsou současně infikováni HIV-1 a HBV, je třeba pečlivě sledovat jaterní funkce s klinickým i laboratorním sledováním po dobu alespoň několika měsíců. Je-li to vhodné, může být opodstatněné zahájení léčby proti hepatitidě B (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ).
Důležité rozdíly mezi přípravky obsahujícími lamivudin
EPIVIR 150 mg tablety a perorální roztok (používaný k léčbě infekce HIV-1) obsahují vyšší dávku léčivé látky (lamivudin) než tablety a perorální roztok EPIVIR-HBV (používané k léčbě chronické infekce HBV). Pacienti s infekcí HIV-1 by měli dostávat pouze lékové formy vhodné pro léčbu HIV-1 (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ).
POPIS
EPIVIR (také známý jako 3TC) je obchodní název pro lamivudin, syntetický nukleosidový analog s aktivitou proti HIV-1 a HBV. Chemický název lamivudinu je (2R,cis)-4-amino-l-(2-hydroxymethyl-l,3-oxathiolan-5-yl)-(lH)-pyrimidin-2-on. Lamivudin je (-)enantiomer dideoxy analogu cytidinu. Lamivudin byl také označován jako (-)2',3'-dideoxy, 3'thiacytidin. Má molekulový vzorec C8H11N3O3S a molekulovou hmotnost 229,3 g na mol. Má následující strukturní vzorec:
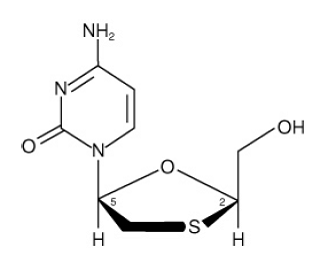
Lamivudin je bílá až téměř bílá krystalická pevná látka s rozpustností přibližně 70 mg na ml ve vodě při 20 °C.
Tablety EPIVIR 150 mg jsou určeny k perorálnímu podání. Každá 150mg potahovaná tableta s půlicí rýhou obsahuje 150 mg lamivudinu a neúčinné složky hypromelózu, magnesium-stearát, mikrokrystalickou celulózu, polyethylenglykol, polysorbát 80, sodnou sůl glykolátu škrobu a oxid titaničitý.
Jedna 300mg potahovaná tableta obsahuje 300 mg lamivudinu a neaktivní složky černý oxid železitý, hypromelózu, magnesium-stearát, mikrokrystalickou celulózu, polyethylenglykol, polysorbát 80, sodnou sůl glykolátu škrobu a oxid titaničitý.
EPIVIR perorální roztok je určen k perorálnímu podání. Jeden mililitr (1 ml) perorálního roztoku EPIVIR obsahuje 10 mg lamivudinu (10 mg na ml) ve vodném roztoku a neaktivní složky umělé jahodové a banánové aroma, kyselina citronová (bezvodá), methylparaben, propylenglykol, propylparaben, citrát sodný (dihydrát) a sacharóza (200 mg).
INDIKACE
EPIVIR 150 mg je nukleosidový analog indikovaný v kombinaci s jinými antiretrovirovými látkami k léčbě infekce virem lidské imunodeficience typu 1 (HIV-1).
Omezení použití
- Dávkování tohoto produktu je pro HIV-1 a ne pro HBV.
DÁVKOVÁNÍ A PODÁVÁNÍ
Doporučené dávkování pro dospělé pacienty
Doporučená dávka přípravku EPIVIR 150 mg u dospělých infikovaných HIV-1 je 300 mg denně, podávaných buď jako 150 mg perorálně dvakrát denně, nebo 300 mg perorálně jednou denně s jídlem nebo bez jídla. Pokud je lamivudin podáván pacientovi infikovanému HIV-1 a HBV, měla by být dávka indikovaná pro léčbu HIV-1 použita jako součást vhodného kombinovaného režimu [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Doporučené dávkování pro pediatrické pacienty
EPIVIR 150 mg tableta s půlicí rýhou je preferovanou formulací pro pediatrické pacienty infikované HIV-1, kteří váží alespoň 14 kg a pro které je vhodná pevná léková forma. Před předepsáním tablet EPIVIR s půlicí rýhou je třeba u pediatrických pacientů vyšetřit schopnost polykat tablety. Pacientům, kteří nejsou schopni bezpečně a spolehlivě spolknout tablety EPIVIR 150 mg, může být předepsána léková forma perorálního roztoku [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]. Doporučená perorální dávka přípravku EPIVIR 150 mg tablety pro pediatrické pacienty infikované HIV-1 je uvedena v tabulce 1.
Orální roztok
Doporučená dávka přípravku EPIVIR 150 mg perorální roztok u pediatrických pacientů infikovaných HIV-1 ve věku 3 měsíců a starších je 5 mg na kg perorálně dvakrát denně nebo 10 mg na kg perorálně jednou denně (až do maximální dávky 300 mg denně), podávané v kombinaci s jinými antiretrovirovými látkami [viz KLINICKÁ FARMAKOLOGIE ]. Při výběru dávkovacího intervalu pro pacienty, kteří zahajují léčbu perorálním roztokem, zvažte virovou zátěž HIV-1 a počet/procento CD4+ buněk [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ , KLINICKÁ FARMAKOLOGIE ].
Pacienti s poruchou ledvin
Dávkování přípravku EPIVIR 150 mg se upravuje podle funkce ledvin. Úpravy dávkování jsou uvedeny v tabulce 2 [viz KLINICKÁ FARMAKOLOGIE ].
Po rutinní (4hodinové) hemodialýze nebo peritoneální dialýze není nutné žádné další dávkování přípravku EPIVIR 150 mg.
Přestože nejsou k dispozici dostatečné údaje pro doporučení specifické úpravy dávky EPIVIRu u pediatrických pacientů s poruchou funkce ledvin, je třeba zvážit snížení dávky a/nebo prodloužení dávkovacího intervalu.
JAK DODÁVÁNO
Dávkové formy A Síly
Tablety EPIVIR
Tablety EPIVIR s rýhou obsahují 150 mg lamivudinu. Tablety jsou bílé potahované tablety ve tvaru kosočtverce s půlicí rýhou a vyraženým „GX CJ7“ na obou stranách.
EPIVIR tablety
Tablety EPIVIR obsahují 300 mg lamivudinu. Tablety jsou šedé, ve tvaru modifikovaného kosočtverce, potažené filmem a s vyrytým „GX EJ7“ na jedné straně a hladké na druhé straně.
EPIVIR perorální roztok
EPIVIR perorální roztok obsahuje 10 mg lamivudinu na 1 ml. Roztok je čirá, bezbarvá až světle žlutá tekutina s příchutí jahod a banánů.
Skladování A Manipulace
EPIVIR Tablety s půlicí rýhou obsahují 150 mg lamivudinu, jsou bílé, ve tvaru kosočtverce, potažené filmem a na obou stranách s vyraženým „GX CJ7“. Baleno takto:
Lahvička 60 tablet ( NDC 49702-203-18) s dětským bezpečnostním uzávěrem.
EPIVIR tablety obsahují 300 mg lamivudinu, jsou šedé, ve tvaru modifikovaného kosočtverce, potažené filmem, s vyrytým „GX EJ7“ na jedné straně a hladké na druhé straně. Baleno takto:
Lahvička 30 tablet ( NDC 49702-204-13) s dětským bezpečnostním uzávěrem.
Doporučené skladování
Tablety EPIVIR skladujte při teplotě 25 °C (77 °F); povoleny výchylky do 15° až 30°C (59° až 86°F) [viz USP řízená pokojová teplota ].
EPIVIR perorální roztok je čirá, bezbarvá až světle žlutá tekutina s příchutí jahody a banánu. Každý ml roztoku obsahuje 10 mg lamivudinu. Baleno takto:
Plastová láhev o objemu 240 ml (NDC 49702-205-48) s dětským bezpečnostním uzávěrem. Tento produkt nevyžaduje rekonstituci.
Uchovávejte v těsně uzavřených lahvích při teplotě 25 ° C (77 ° F) [viz USP řízená pokojová teplota ].
Vyrobeno pro: ViiV Healthcare Research Triangle Park, NC 27709. Autor: GlaxoSmithKline Research Triangle Park, NC 27709, Vyrobeno se souhlasem Shire Pharmaceuticals Group plc. Revize: duben 2018
VEDLEJŠÍ EFEKTY
Následující nežádoucí účinky jsou popsány v jiných částech označení:
- Exacerbace hepatitidy B [viz VAROVÁNÍ V KRABICE , VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
- Laktátová acidóza a těžká hepatomegalie se steatózou [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
- Dekompenzace jater u pacientů koinfikovaných HIV-1 a hepatitidou C [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
- Pankreatitida [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
- Imunitní rekonstituční syndrom [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Zkušenosti z klinických studií
Zkušenosti z klinických studií u dospělých subjektů
Vzhledem k tomu, že klinické studie probíhají za velmi odlišných podmínek, nelze míry nežádoucích reakcí pozorované v klinických studiích léku přímo srovnávat s mírami v klinických studiích jiného léku a nemusí odrážet míry pozorované v klinické praxi.
Bezpečnostní profil přípravku EPIVIR 150 mg u dospělých je primárně založen na 3 568 jedincích infikovaných HIV-1 v 7 klinických studiích.
Nejčastějšími nežádoucími účinky jsou bolest hlavy, nevolnost, malátnost, únava, nosní příznaky a symptomy, průjem a kašel.
Vybrané klinické nežádoucí účinky u více než nebo rovných 5 % subjektů během léčby přípravkem EPIVIR 150 mg dvakrát denně plus RETROVIR 200 mg 3krát denně po dobu až 24 týdnů jsou uvedeny v tabulce 3.
Pankreatitida
Pankreatitida byla pozorována u 9 z 2 613 dospělých subjektů (0,3 %), kteří dostávali EPIVIR 150 mg v kontrolovaných klinických studiích EPV20001, NUCA3001, NUCB3001, NUCA3002, NUCB3002 a NUCB3007 [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
EPIVIR 300 mg jednou denně
Typy a frekvence klinických nežádoucích účinků hlášených u subjektů užívajících EPIVIR 300 mg jednou denně nebo EPIVIR 150 mg dvakrát denně (v režimech s kombinací 3 léčiv v EPV20001 a EPV40001) po dobu 48 týdnů byly podobné.
Vybrané laboratorní abnormality pozorované během terapie jsou shrnuty v tabulce 4.
Frekvence vybraných laboratorních abnormalit hlášených u subjektů užívajících EPIVIR 300 mg jednou denně nebo EPIVIR 150 mg dvakrát denně (v režimech 3 kombinací léků v EPV20001 a EPV40001) byly podobné.
Zkušenosti z klinických studií u pediatrických subjektů
EPIVIR perorální roztok byl studován u 638 pediatrických pacientů ve věku od 3 měsíců do 18 let ve 3 klinických studiích.
Vybrané klinické nežádoucí účinky a fyzikální nálezy s četností vyšší nebo rovnou 5 % během léčby přípravkem EPIVIR 4 mg na kg dvakrát denně plus RETROVIR 160 mg na m² 3krát denně u dosud neléčených (méně než nebo rovných 56 dnům antiretrovirové léčby terapie) dětské subjekty jsou uvedeny v tabulce 5.
Pankreatitida
Pankreatitida, která byla v některých případech fatální, byla pozorována u pediatrických pacientů již dříve léčených antiretrovirovými nukleosidy, kteří dostávali EPIVIR samotný nebo v kombinaci s jinými antiretrovirovými přípravky. V otevřené studii s eskalací dávky (NUCA2002) se u 14 subjektů (14 %) vyvinula pankreatitida při monoterapii EPIVIRem. Tři z těchto subjektů zemřeli na komplikace pankreatitidy. Ve druhé otevřené studii (NUCA2005) se u 12 subjektů (18 %) vyvinula pankreatitida. Ve studii ACTG300 nebyla pankreatitida pozorována u 236 subjektů randomizovaných do EPIVIR 150 mg plus RETROVIR. Pankreatitida byla pozorována u 1 subjektu v této studii, který dostával otevřený EPIVIR v kombinaci s RETROVIREM a ritonavirem po přerušení monoterapie didanosinem (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Parestézie a periferní neuropatie
Parestézie a periferní neuropatie byly hlášeny u 15 subjektů (15 %) ve studii NUCA2002, 6 subjektů (9 %) ve studii NUCA2005 a 2 subjektů (méně než 1 %) ve studii ACTG300.
Vybrané laboratorní abnormality zaznamenané u pediatrických pacientů dosud neléčených (méně než nebo rovnající se 56 dnům antiretrovirové terapie) jsou uvedeny v tabulce 6.
Pediatričtí jedinci jednou denně versus dávkování dvakrát denně (COL105677)
Bezpečnost podávání jednou denně ve srovnání s dávkováním EPIVIR 150 mg dvakrát denně byla hodnocena ve studii ARROW. Primární hodnocení bezpečnosti ve studii ARROW bylo založeno na nežádoucích účincích 3. a 4. stupně. Frekvence nežádoucích účinků stupně 3 a 4 byla podobná u subjektů randomizovaných k podávání jednou denně ve srovnání se subjekty randomizovanými k podávání dávky dvakrát denně. Jedna událost hepatitidy 4. stupně v kohortě užívající jednou denně byla zkoušejícím považována za nejistou kauzalitu a všechny ostatní nežádoucí účinky 3. nebo 4. stupně byly zkoušejícím považovány za nesouvisející.
Novorozenci
Omezené informace o krátkodobé bezpečnosti jsou dostupné ze 2 malých, nekontrolovaných studií v Jižní Africe u novorozenců užívajících lamivudin se zidovudinem nebo bez něj po první týden života po léčbě matky počínaje 38. nebo 36. týdnem těhotenství [viz KLINICKÁ FARMAKOLOGIE ]. Vybrané nežádoucí účinky hlášené u těchto novorozenců zahrnovaly zvýšené jaterní testy, anémii, průjem, poruchy elektrolytů, hypoglykémii, žloutenku a hepatomegalii, vyrážku, respirační infekce a sepsi; 3 novorozenci zemřeli (1 na gastroenteritidu s acidózou a křečemi, 1 na traumatické poranění a 1 z neznámých příčin). Byly hlášeny dva další nefatální případy gastroenteritidy nebo průjmu, včetně 1 s křečemi; 1 kojenec měl přechodnou renální insuficienci spojenou s dehydratací. Absence kontrolních skupin omezuje hodnocení kauzality, ale mělo by se předpokládat, že perinatálně exponovaní kojenci mohou mít riziko nežádoucích účinků srovnatelné s těmi, které byly hlášeny u pediatrických a dospělých pacientů infikovaných HIV-1 léčených kombinovanými režimy obsahujícími lamivudin. Dlouhodobé účinky expozice lamivudinu in utero a kojenců nejsou známy.
Postmarketingové zkušenosti
Následující nežádoucí účinky byly identifikovány během používání EPIVIRu po schválení. Protože jsou tyto reakce hlášeny dobrovolně z populace neznámé velikosti, není vždy možné spolehlivě odhadnout jejich frekvenci nebo stanovit příčinnou souvislost s expozicí léku. Tyto reakce byly vybrány pro zařazení kvůli kombinaci jejich závažnosti, četnosti hlášení nebo potenciální kauzální souvislosti s lamivudinem.
Tělo jako celek
Redistribuce/akumulace tělesného tuku.
Endokrinní a metabolické
Hyperglykémie.
Všeobecné
Slabost. Hemická a lymfatická anémie (včetně čisté aplazie červených krvinek a závažných anémií progredujících během léčby).
Jaterní a pankreatický
Laktátová acidóza a steatóza jater [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ], exacerbace hepatitidy B po léčbě [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Přecitlivělost
Anafylaxe, kopřivka.
Muskuloskeletální
Svalová slabost, elevace CPK, rhabdomyolýza. Kožní alopecie, pruritus.
DROGOVÉ INTERAKCE
Léky inhibující transportéry organických kationtů
Lamivudin je vylučován převážně močí aktivní organickou kationtovou sekrecí. Měla by být zvážena možnost interakcí s jinými souběžně podávanými léky, zejména pokud je jejich hlavní cestou eliminace aktivní renální sekrece prostřednictvím organického kationtového transportního systému (např. trimethoprim) [viz KLINICKÁ FARMAKOLOGIE ]. Nejsou k dispozici žádné údaje o interakcích s jinými léky, které mají mechanismus renální clearance podobný mechanismu lamivudinu.
Sorbitol
Současné podávání jednotlivých dávek lamivudinu a sorbitolu vedlo ke snížení expozice lamivudinu závislému na dávce sorbitolu. Pokud je to možné, vyhněte se užívání léků obsahujících sorbitol s lamivudinem (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ , KLINICKÁ FARMAKOLOGIE ].
VAROVÁNÍ
Zahrnuto jako součást OPATŘENÍ sekce.
OPATŘENÍ
Pacienti s koinfekcí virem hepatitidy B
Exacerbace hepatitidy po léčbě
Po vysazení lamivudinu se objevily klinické a laboratorní známky exacerbací hepatitidy. Tyto exacerbace byly kromě opětovného výskytu HBV DNA detekovány především zvýšením sérové ALT. Ačkoli se zdá, že většina příhod byla omezena sama na sebe, v některých případech byly hlášeny smrtelné případy. Podobné příhody byly hlášeny z postmarketingových zkušeností po změnách z léčebných režimů obsahujících lamivudin HIV-1 na režimy bez lamivudinu u pacientů infikovaných HIV-1 i HBV. Příčinná souvislost s přerušením léčby lamivudinem není známa. Pacienti by měli být pečlivě sledováni klinicky i laboratorně po dobu alespoň několika měsíců po ukončení léčby.
Důležité rozdíly mezi produkty obsahujícími lamivudin
EPIVIR 150 mg tablety a perorální roztok obsahují vyšší dávku stejné účinné látky (lamivudin) než tablety EPIVIR-HBV a perorální roztok EPIVIR-HBV. EPIVIR-HBV byl vyvinut pro pacienty s chronickou hepatitidou B. Formulace a dávkování lamivudinu v EPIVIR-HBV nejsou vhodné pro pacienty koinfikované HIV-1 a HBV. Bezpečnost a účinnost lamivudinu pro léčbu chronické hepatitidy B u pacientů současně infikovaných HIV-1 a HBV nebyla stanovena. Pokud je léčba EPIVIR-HBV předepsána pro chronickou hepatitidu B u pacienta s nerozpoznanou nebo neléčenou infekcí HIV-1, pravděpodobně dojde k rychlému vzniku rezistence na HIV-1 kvůli subterapeutické dávce a nevhodnosti monoterapie léčby HIV-1. Pokud se rozhodne podávat lamivudin pacientům současně infikovaným HIV-1 a HBV, je třeba jako součást vhodného kombinovaného režimu použít EPIVIR 150 mg tablety, EPIVIR 150 mg perorální roztok nebo jiný přípravek obsahující vyšší dávku lamivudinu.
Vznik HBV rezistentní na lamivudin
Bezpečnost a účinnost lamivudinu pro léčbu chronické hepatitidy B u subjektů duálně infikovaných HIV-1 a HBV nebyla stanovena (viz úplné informace o předepisování EPIVIR-HBV). Vznik variant viru hepatitidy B spojený s rezistencí na lamivudin byl také hlášen u subjektů infikovaných HIV-1, kteří dostávali antiretrovirové režimy obsahující lamivudin v přítomnosti souběžné infekce virem hepatitidy B.
Laktátová acidóza a těžká hepatomegalie se steatózou
Laktátová acidóza a těžká hepatomegalie se steatózou, včetně fatálních případů, byly hlášeny při použití nukleosidových analogů, včetně EPIVIR. Většina těchto případů byla u žen. Ženské pohlaví a obezita mohou být rizikovými faktory pro rozvoj laktátové acidózy a těžké hepatomegalie se steatózou u pacientů léčených antiretrovirovými nukleosidovými analogy. Léčba přípravkem EPIVIR by měla být přerušena u každého pacienta, u kterého se rozvinou klinické nebo laboratorní nálezy naznačující laktátovou acidózu nebo výraznou hepatotoxicitu, která může zahrnovat hepatomegalii a steatózu, a to i při absenci výrazného zvýšení transamináz.
Použití s režimy na bázi interferonu a ribavirinu
Studie in vitro ukázaly, že ribavirin může snížit fosforylaci pyrimidinových nukleosidových analogů, jako je lamivudin. Ačkoli nebyly pozorovány žádné známky farmakokinetické nebo farmakodynamické interakce (např. ztráta virologické suprese HIV-1/HCV), když byl ribavirin podáván současně s lamivudinem u pacientů koinfikovaných HIV-1/HCV (viz KLINICKÁ FARMAKOLOGIE došlo k dekompenzaci jater (některé fatální) u pacientů koinfikovaných HIV-1/HCV, kteří dostávali kombinovanou antiretrovirovou terapii HIV-1 a interferonu alfa s ribavirinem nebo bez ribavirinu. Pacienti užívající interferon alfa s ribavirinem nebo bez ribavirinu a EPIVIR by měli být pečlivě sledováni z hlediska toxicity souvisejících s léčbou, zejména jaterní dekompenzace. Ukončení léčby přípravkem EPIVIR 150 mg by mělo být považováno za lékařsky vhodné. Pokud je pozorována zhoršující se klinická toxicita, včetně jaterní dekompenzace (např. Child-Pugh vyšší než 6), je třeba zvážit snížení dávky nebo vysazení interferonu alfa, ribavirinu nebo obou. Viz úplné informace o předepisování interferonu a ribavirinu.
Pankreatitida
pediatrických pacientů s anamnézou předchozí expozice antiretrovirovým nukleosidům, anamnézou pankreatitidy nebo jinými významnými rizikovými faktory pro rozvoj pankreatitidy by měl být EPIVIR 150 mg používán s opatrností. Léčba přípravkem EPIVIR 150 mg by měla být okamžitě ukončena, pokud se objeví klinické známky, příznaky nebo laboratorní abnormality svědčící pro pankreatitidu (viz NEŽÁDOUCÍ REAKCE ].
Syndrom imunitní rekonstituce
U pacientů léčených kombinovanou antiretrovirovou terapií, včetně EPIVIR, byl hlášen syndrom imunitní rekonstituce. Během počáteční fáze kombinované antiretrovirové léčby se u pacientů, jejichž imunitní systém reaguje, může vyvinout zánětlivá odpověď na indolentní nebo reziduální oportunní infekce (jako je infekce Mycobacterium avium, cytomegalovirus, pneumonie způsobená Pneumocystis jirovecii [PCP] nebo tuberkulóza), což může vyžadovat další vyšetření a léčbu.
Autoimunitní poruchy (jako je Gravesova choroba, polymyositida a Guillain-Barrého syndrom) byly také hlášeny v souvislosti s rekonstitucí imunity, avšak doba do nástupu je variabilnější a může se objevit mnoho měsíců po zahájení léčby.
Nižší míra virologické suprese a zvýšené riziko virové rezistence s perorálním roztokem
Pediatričtí pacienti, kteří dostávali EPIVIR 150 mg perorální roztok (v dávkách na základě hmotnostního pásma přibližně 8 mg na kg za den) současně s jinými antiretrovirovými perorálními roztoky kdykoli ve studii ARROW, měli nižší míru virologické suprese, nižší plazmatickou expozici lamivudinu a vyvinuli virová rezistence častěji než ti, kteří dostávají tablety EPIVIR [viz KLINICKÁ FARMAKOLOGIE , Mikrobiologie , Klinické studie ].
Tableta EPIVIR s rýhou je upřednostňovanou formulací pro pediatrické pacienty infikované HIV-1, kteří váží alespoň 14 kg a pro které je vhodná pevná léková forma. Pokud je to možné, je třeba použít režim všech tablet, aby se zabránilo potenciální interakci se sorbitolem [viz KLINICKÁ FARMAKOLOGIE ]. Při léčbě přípravkem EPIVIR 150 mg perorální roztok zvažte častější monitorování virové zátěže HIV-1.
Informace pro pacienty
Doporučte pacientovi, aby si přečetl označení pacienta schválené FDA (Informace pro pacienta).
Pacienti se souběžnou infekcí hepatitidou B nebo C
Informujte pacienty koinfikované HIV-1 a HBV, že při přerušení léčby lamivudinem v některých případech došlo ke zhoršení jaterního onemocnění. Poraďte pacientům, aby jakékoli změny v režimu prodiskutovali se svým poskytovatelem zdravotní péče [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Informujte pacienty s koinfekcí HIV-1/HCV, že u pacientů koinfikovaných HIV-1/HCV, kteří dostávali kombinovanou antiretrovirovou terapii pro HIV-1 a interferon alfa s ribavirinem nebo bez ribavirinu, došlo k jaterní dekompenzaci (některé fatální) [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Rozdíly ve formulacích EPIVIR
Informujte pacienty, že EPIVIR 150 mg tablety a perorální roztok obsahují vyšší dávku stejné účinné látky (lamivudin) jako tablety a perorální roztok EPIVIR-HBV. Pokud je učiněno rozhodnutí zařadit lamivudin do léčebného režimu HIV-1 pacienta koinfikovaného HIV-1 a HBV, měla by být použita formulace a dávkování lamivudinu v EPIVIR (nikoli EPIVIR-HBV) [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Laktátová acidóza/hepatomegalie se steatózou
Upozorněte pacienty, že při užívání nukleosidových analogů a jiných antiretrovirálních léků byla hlášena laktátová acidóza a těžká hepatomegalie se steatózou. Informujte pacienty, aby přestali užívat EPIVIR, pokud se u nich rozvinou klinické příznaky naznačující laktátovou acidózu nebo výraznou hepatotoxicitu (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Riziko pankreatitidy
Poraďte rodičům nebo opatrovníkům, aby sledovali dětské pacienty na známky a příznaky pankreatitidy [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Syndrom imunitní rekonstituce
Informujte pacienty, aby okamžitě informovali svého poskytovatele zdravotní péče o jakýchkoli známkách a příznacích infekce, protože brzy po kombinované antiretrovirové léčbě se může objevit zánět z předchozí infekce, včetně zahájení léčby přípravkem EPIVIR [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Nižší míra virologické suprese a zvýšené riziko virové rezistence s perorálním roztokem
Informujte pacienty, že pokud je to možné, měl by být použit režim všech tablet kvůli zvýšené míře selhání léčby u pediatrických pacientů, kteří dostávali perorální roztok EPIVIR současně s jinými antiretrovirovými perorálními roztoky (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Obsah sacharózy v perorálním roztoku EPIVIR
Informujte diabetické pacienty, že každá 15ml dávka perorálního roztoku EPIVIR obsahuje 3 gramy sacharózy (1 ml = 200 mg sacharózy) [viz POPIS ].
Registr těhotenství
Informujte pacienty, že existuje registr těhotenských expozic, který monitoruje výsledky těhotenství u žen vystavených EPIVIRu během těhotenství [viz Použití u konkrétních populací ].
Laktace
Poučte ženy s infekcí HIV-1, aby nekojily, protože HIV-1 se může přenést na dítě mateřským mlékem (viz Použití u konkrétních populací ].
Zmeškané dávkování
Poučte pacienty, že pokud vynechají dávku přípravku EPIVIR 150 mg, aby ji užili, jakmile si vzpomenou. Informujte pacienty, aby nezdvojnásobovali svou další dávku nebo neužívali více než předepsanou dávku [viz DÁVKOVÁNÍ A PODÁVÁNÍ ].
EPIVIR a RETROVIR jsou ochranné známky vlastněné nebo licencované skupinou společností ViiV Healthcare.
Neklinická toxikologie
Karcinogeneze, mutageneze, zhoršení plodnosti
Karcinogeneze
Dlouhodobé studie karcinogenity s lamivudinem u myší a potkanů neprokázaly žádný důkaz karcinogenního potenciálu při expozicích až 10násobku (myši) a 58násobku (potkani) expozice u člověka při doporučené dávce 300 mg.
Mutageneze
Lamivudin byl mutagenní v testu myšího lymfomu L5178Y a klastogenní v cytogenetickém testu s použitím kultivovaných lidských lymfocytů. Lamivudin nebyl mutagenní v testu mikrobiální mutagenity, v testu buněčné transformace in vitro, v mikronukleárním testu na potkanech, v cytogenetickém testu kostní dřeně potkanů a ve stanovení neplánované syntézy DNA v játrech potkana. Lamivudin nevykazoval žádné známky genotoxické aktivity in vivo u potkanů při perorálních dávkách až 2 000 mg na kg, které produkovaly plazmatické hladiny 35 až 45krát vyšší než u lidí při dávce doporučené pro infekci HIV-1.
Zhoršení Plodnosti
Ve studii reprodukční schopnosti lamivudin podávaný potkanům v dávkách až 4 000 mg na kg za den, produkující plazmatické hladiny 47 až 70krát vyšší než u lidí, neodhalil žádné známky poškození plodnosti a žádný vliv na přežití, růst a vývoj k odstavení potomstva.
Použití u konkrétních populací
Těhotenství
Registr expozice v těhotenství
Existuje registr těhotenských expozic, který monitoruje výsledky těhotenství u žen vystavených EPIVIRu během těhotenství. Poskytovatelům zdravotní péče se doporučuje, aby registrovali pacienty zavoláním do registru antiretrovirových těhotenství (APR) na čísle 1-800-258-4263.
Shrnutí rizik
Dostupné údaje z APR neukazují žádný rozdíl v celkovém riziku vrozených vad u lamivudinu ve srovnání se základní mírou vrozených vad 2,7 % v referenční populaci Metropolitan Atlanta Congenital Defects Program (MACDP) (viz Data ). APR používá MACDP jako referenční populaci USA pro vrozené vady v běžné populaci. MACDP hodnotí ženy a kojence z omezené geografické oblasti a nezahrnuje výsledky pro porody, ke kterým došlo v době méně než 20 týdnů těhotenství. Míra potratů se v RPSN neuvádí. Odhadovaná základní míra potratů u klinicky uznaných těhotenství v běžné populaci USA je 15 % až 20 %. Základní riziko závažných vrozených vad a potratu pro indikovanou populaci není známo.
V reprodukčních studiích na zvířatech vedlo perorální podávání lamivudinu březím králíkům během organogeneze k embryoletalitě při systémové expozici (AUC) podobné doporučené klinické dávce; nebyly však pozorovány žádné nežádoucí účinky na vývoj při perorálním podávání lamivudinu březím potkanům během organogeneze při plazmatických koncentracích (Cmax) 35násobku doporučené klinické dávky (viz Data ).
Data
Lidská data
Na základě prospektivních zpráv k APR o více než 11 000 expozicích lamivudinu během těhotenství, které vedly k živě narozeným dětem (včetně více než 4 500 vystavených v prvním trimestru), nebyl žádný rozdíl mezi celkovým rizikem vrozených vad u lamivudinu ve srovnání se základní mírou vrozených vad 2,7 % v americké referenční populaci MACDP. Prevalence defektů u živě narozených dětí byla 3,1 % (95% CI: 2,6 % až 3,6 %) po expozici v prvním trimestru režimem obsahujícím lamivudin a 2,8 % (95 % CI: 2,5 % až 3,3 %) po expozici ve druhém/třetím trimestru na režimy obsahující lamivudin.
Farmakokinetika lamivudinu byla studována u těhotných žen během 2 klinických studií provedených v Jižní Africe. Ve studiích byla hodnocena farmakokinetika u 16 žen ve 36. týdnu těhotenství s použitím 150 mg lamivudinu dvakrát denně se zidovudinem, u 10 žen ve 38. týdnu těhotenství s použitím 150 mg lamivudinu dvakrát denně se zidovudinem a u 10 žen ve 38. týdnu těhotenství s použitím 300 mg lamivudinu dvakrát denně bez dalších antiretrovirotik. Tyto studie nebyly navrženy ani poháněny tak, aby poskytovaly informace o účinnosti. Koncentrace lamivudinu byly obecně podobné ve vzorcích mateřského, neonatálního a pupečníkového séra. U podskupiny subjektů byly vzorky plodové vody odebrány po přirozené ruptuře membrán a potvrdilo se, že lamivudin u lidí prochází placentou. Na základě omezených údajů při porodu byly střední (rozsah) koncentrací lamivudinu v plodové vodě 3,9 (1,2 až 12,8)–krát vyšší ve srovnání s párovou koncentrací v mateřském séru (n = 8).
Údaje o zvířatech
Lamivudin byl podáván perorálně březím potkanům (v dávkách 90, 600 a 4 000 mg na kg za den) a králíkům (v dávkách 90, 300 a 1 000 mg na kg za den a v dávkách 15, 40 a 90 mg na kg za den) během organogeneze (7 až 16. březost [potkan] a 8 až 20 [králík]). U potkanů a králíků nebyly pozorovány žádné známky malformací plodu způsobené lamivudinem při dávkách produkujících plazmatické koncentrace (Cmax) přibližně 35krát vyšší, než je expozice u člověka při doporučené denní dávce. Důkaz časné embryoletality byl pozorován u králíků při systémových expozicích (AUC) podobných těm, které byly pozorovány u lidí, ale u potkanů nebyly žádné známky tohoto účinku při plazmatických koncentracích (Cmax) 35krát vyšších než je expozice u člověka při doporučené denní dávce . Studie na březích potkaních potkanech ukázaly, že lamivudin se do plodu přenáší přes placentu. Ve studii fertility/pre- a postnatálního vývoje u potkanů byl lamivudin podáván perorálně v dávkách 180, 900 a 4 000 mg na kg za den (od před pářením do postnatálního dne 20). Ve studii nebyl vývoj potomstva, včetně fertility a reprodukční výkonnosti, ovlivněn podáváním lamivudinu matce.
Laktace
Shrnutí rizik
Centrum pro kontrolu a prevenci nemocí doporučuje, aby matky infikované HIV-1 ve Spojených státech nekojily své děti, aby se vyhnuly riziku postnatálního přenosu infekce HIV-1. Lamivudin je přítomen v lidském mléce. Neexistují žádné informace o účincích lamivudinu na kojené dítě nebo o účincích léků na tvorbu mléka. Z důvodu možného (1) přenosu HIV-1 (u HIV-negativních kojenců), (2) rozvoje virové rezistence (u HIV-pozitivních dětí) a (3) nežádoucích reakcí u kojeného dítěte poučte matky, aby nekojily pokud dostávají EPIVIR.
Pediatrické použití
Bezpečnost a účinnost přípravku EPIVIR 150 mg v kombinaci s jinými antiretrovirovými látkami byla stanovena u pediatrických pacientů ve věku 3 měsíců a starších. Tableta EPIVIR s rýhou je preferovanou formulací pro pediatrické pacienty infikované HIV-1, kteří váží alespoň 14 kg a pro které je vhodná pevná léková forma, protože dětští pacienti, kteří dostávali EPIVIR 150 mg perorální roztok, měli nižší míru virologické suprese, nižší plazmatickou expozici lamivudinu a vyvinula se u nich virová rezistence častěji než u pacientů, kteří dostávali tablety EPIVIR 150 mg ve studii ARROW [viz DÁVKOVÁNÍ A PODÁVÁNÍ , VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ , NEŽÁDOUCÍ REAKCE , KLINICKÁ FARMAKOLOGIE , Klinické studie ].
Geriatrické použití
Klinické studie EPIVIR nezahrnovaly dostatečný počet subjektů ve věku 65 let a více, aby bylo možné určit, zda reagují odlišně od mladších subjektů. Obecně je třeba dbát opatrnosti při podávání EPIVIR u starších pacientů, což odráží vyšší frekvenci snížené funkce jater, ledvin nebo srdce a souběžného onemocnění nebo jiné lékové terapie (viz DÁVKOVÁNÍ A PODÁVÁNÍ , KLINICKÁ FARMAKOLOGIE ].
Pacienti s poruchou funkce ledvin
U pacientů s poruchou funkce ledvin se doporučuje snížení dávky EPIVIR 150 mg (viz DÁVKOVÁNÍ A PODÁVÁNÍ , KLINICKÁ FARMAKOLOGIE ].
PŘEDÁVKOVAT
Není známa žádná specifická léčba předávkování přípravkem EPIVIR. Pokud dojde k předávkování, pacient by měl být sledován a podle potřeby mu musí být aplikována standardní podpůrná léčba. Protože zanedbatelné množství lamivudinu bylo odstraněno pomocí (4hodinové) hemodialýzy, kontinuální ambulantní peritoneální dialýzy a automatizované peritoneální dialýzy, není známo, zda by kontinuální hemodialýza poskytla klinický přínos v případě předávkování lamivudinem.
KONTRAINDIKACE
EPIVIR 150 mg je kontraindikován u pacientů s předchozí hypersenzitivní reakcí na lamivudin.
KLINICKÁ FARMAKOLOGIE
Mechanismus působení
Lamivudin je antiretrovirová látka [viz Mikrobiologie ].
Farmakokinetika
Farmakokinetika u dospělých
Farmakokinetické vlastnosti lamivudinu byly studovány u asymptomatických dospělých jedinců infikovaných HIV-1 po podání jednotlivých intravenózních (IV) dávek v rozmezí od 0,25 do 8 mg na kg, jakož i jednorázových a opakovaných (v režimu dvakrát denně) perorálních dávek v rozmezí od 0,25 do 10 mg na kg.
Farmakokinetické vlastnosti lamivudinu byly také studovány jako jednorázové a opakované perorální dávky v rozmezí od 5 mg do 600 mg denně podávané subjektům infikovaným HBV.
Farmakokinetické vlastnosti tablety EPIVIR 300 mg jednou denně po dobu 7 dnů v ustáleném stavu ve srovnání s tabletou EPIVIR 150 mg dvakrát denně po dobu 7 dnů byly hodnoceny ve zkřížené studii u 60 zdravých subjektů. EPIVIR 300 mg jednou denně vedl k expozicím lamivudinu, které byly podobné jako u EPIVIRu 150 mg dvakrát denně s ohledem na plazmatickou AUC24,ss; avšak Cmax,ss byla o 66 % vyšší a minimální hodnota byla o 53 % nižší ve srovnání s režimem 150 mg dvakrát denně. Intracelulární expozice lamivudintrifosfátu v mononukleárních buňkách periferní krve byly také podobné, pokud jde o AUC24,ss a Cmax24,ss; minimální hodnoty však byly nižší ve srovnání s režimem 150 mg dvakrát denně. Interindividuální variabilita byla větší u intracelulárních koncentrací lamivudintrifosfátu oproti minimálním plazmatickým koncentracím lamivudinu.

Farmakokinetika lamivudinu byla hodnocena u 12 dospělých jedinců infikovaných HIV-1, kterým byl podáván lamivudin v dávce 150 mg dvakrát denně v kombinaci s jinými antiretrovirovými přípravky. Geometrický průměr (95% CI) pro AUC(0-12) byl 5,53 (4,58, 6,67) mcg.h na ml a pro Cmax byl 1,40 (1,17, 1,69) mcg na ml.
Absorpce a biologická dostupnost
Absolutní biologická dostupnost u 12 dospělých subjektů byla 86 % ± 16 % (průměr ± SD) pro 150mg tabletu a 87 % ± 13 % pro perorální roztok. Po perorálním podávání 2 mg na kg dvakrát denně 9 dospělým s HIV-1 byla maximální koncentrace lamivudinu v séru (Cmax) 1,5 ± 0,5 mcg na ml (průměr ± SD). Plocha pod křivkou plazmatické koncentrace versus čas (AUC) a Cmax se zvyšovaly úměrně k perorální dávce v rozmezí od 0,25 do 10 mg na kg.
Poměr akumulace lamivudinu u HIV-1-pozitivních asymptomatických dospělých s normální funkcí ledvin byl 1,50 po 15 dnech perorálního podávání 2 mg na kg dvakrát denně.
Účinky Potraviny Na Orální Absorpci
EPIVIR 150 mg tablety a perorální roztok lze podávat s jídlem nebo bez jídla. Zkoumaná 25mg léková forma lamivudinu byla podána perorálně 12 asymptomatickým subjektům infikovaným HIV-1 dvakrát, jednou nalačno a jednou s jídlem (1 099 kcal; 75 gramů tuku, 34 gramů bílkovin, 72 gramů sacharidů ). Absorpce lamivudinu byla pomalejší ve stavu po jídle (Tmax: 3,2 ± 1,3 hodiny) ve srovnání se stavem nalačno (Tmax: 0,9 ± 0,3 hodiny); Cmax ve stavu nasycení byla o 40 % ± 23 % (průměr ± SD) nižší než ve stavu nalačno. Nebyl žádný významný rozdíl v systémové expozici (AUC∞) ve stavu nasycení a nalačno.
Rozdělení
Zdánlivý distribuční objem po IV podání lamivudinu 20 subjektům byl 1,3 ± 0,4 l/kg, což naznačuje, že lamivudin se distribuuje do extravaskulárních prostor. Distribuční objem byl nezávislý na dávce a nekoreloval s tělesnou hmotností.
Vazba lamivudinu na lidské plazmatické proteiny je nižší než 36 %. Studie in vitro ukázaly, že v rozmezí koncentrací 0,1 až 100 mcg na ml se množství lamivudinu spojeného s erytrocyty pohybovalo od 53 % do 57 % a bylo nezávislé na koncentraci.
Metabolismus
Metabolismus lamivudinu je vedlejší cestou eliminace. U lidí je jediným známým metabolitem lamivudinu trans-sulfoxidový metabolit (přibližně 5 % perorální dávky po 12 hodinách). Sérové koncentrace tohoto metabolitu nebyly stanoveny. Lamivudin není významně metabolizován enzymy cytochromu P450.
Odstranění
Většina lamivudinu je eliminována v nezměněné podobě močí aktivní organickou kationtovou sekrecí. U 9 zdravých subjektů, kterým byla podána jedna 300mg perorální dávka lamivudinu, byla renální clearance 199,7 ± 56,9 ml/min (průměr ± SD). U 20 jedinců infikovaných HIV-1, kterým byla podána jednorázová IV dávka, byla renální clearance 280,4 ± 75,2 ml/min (průměr ± SD), což představuje 71 % ± 16 % (průměr ± SD) celkové clearance lamivudinu.
Ve většině studií s jednorázovou dávkou u jedinců infikovaných HIV-1, jedinců infikovaných HBV nebo zdravých jedinců s odběrem séra po dobu 24 hodin po podání dávky se pozorovaný průměrný poločas eliminace (t½) pohyboval v rozmezí 5 až 7 hodin. U jedinců infikovaných HIV-1 byla celková clearance 398,5 ± 69,1 ml za minutu (průměr ± SD). Perorální clearance a eliminační poločas byly nezávislé na dávce a tělesné hmotnosti v rozmezí perorálního dávkování 0,25 až 10 mg na kg.
Specifické populace
Pacienti s poruchou ledvin
Farmakokinetické vlastnosti lamivudinu byly stanoveny u malé skupiny dospělých infikovaných HIV-1 s poruchou funkce ledvin (tabulka 7).
Tmax nebyl významně ovlivněn funkcí ledvin. Na základě těchto pozorování se doporučuje upravit dávkování lamivudinu u pacientů s poruchou funkce ledvin (viz DÁVKOVÁNÍ A PODÁVÁNÍ ].
Na základě studie u jinak zdravých jedinců s poruchou funkce ledvin zvýšila hemodialýza clearance lamivudinu z průměrných 64 na 88 ml/min; délka hemodialýzy (4 hodiny) však nebyla dostatečná k tomu, aby významně změnila průměrnou expozici lamivudinu po podání jedné dávky. Kontinuální ambulantní peritoneální dialýza a automatizovaná peritoneální dialýza mají zanedbatelný vliv na clearance lamivudinu. Proto se po úpravě dávky na clearance kreatininu doporučuje neprovádět žádné další úpravy dávky po rutinní hemodialýze nebo peritoneální dialýze.
Účinky poruchy funkce ledvin na farmakokinetiku lamivudinu u pediatrických pacientů nejsou známy.
Pacienti s poruchou funkce jater
Farmakokinetické vlastnosti lamivudinu byly stanoveny u dospělých s poruchou funkce jater. Farmakokinetické parametry nebyly změněny sníženou funkcí jater. Bezpečnost a účinnost lamivudinu nebyla stanovena v přítomnosti dekompenzovaného onemocnění jater.
Těhotná žena
Farmakokinetika lamivudinu byla studována u 36 těhotných žen během 2 klinických studií provedených v Jižní Africe. Farmakokinetika lamivudinu u těhotných žen byla podobná jako u netěhotných dospělých a u žen po porodu. Koncentrace lamivudinu byly obecně podobné ve vzorcích mateřského, neonatálního a pupečníkového séra.
Pediatričtí pacienti
Farmakokinetika lamivudinu byla studována po jednorázovém nebo opakovaném podání EPIVIRu u 210 pediatrických pacientů. Pediatričtí pacienti užívající perorální roztok lamivudinu (v dávce přibližně 8 mg na kg denně) dosáhli přibližně o 25 % nižších plazmatických koncentrací lamivudinu ve srovnání s dospělými infikovanými HIV-1. Pediatričtí pacienti užívající lamivudin perorální tablety dosáhli plazmatických koncentrací srovnatelných nebo mírně vyšších než koncentrace pozorované u dospělých. Absolutní biologická dostupnost tablet EPIVIR i perorálního roztoku je u dětí nižší než u dospělých. Relativní biologická dostupnost perorálního roztoku EPIVIR je u pediatrických pacientů přibližně o 40 % nižší než u tablet obsahujících lamivudin, navzdory žádnému rozdílu u dospělých. Nižší expozice lamivudinu u pediatrických pacientů užívajících EPIVIR 150 mg perorální roztok je pravděpodobně způsobena interakcí mezi lamivudinem a současně podávanými roztoky obsahujícími sorbitol (jako je ZIAGEN). Modelování farmakokinetických údajů naznačuje, že k dosažení dostatečných koncentrací lamivudinu je zapotřebí zvýšení dávky perorálního roztoku EPIVIR na 5 mg na kg užívaného perorálně dvakrát denně nebo 10 mg na kg užívaného perorálně jednou denně (až do maxima 300 mg denně) [viz DÁVKOVÁNÍ A PODÁVÁNÍ ]. Neexistují žádné klinické údaje u pediatrických pacientů infikovaných HIV-1, kteří byli v této dávce současně podáváni s léky obsahujícími sorbitol.
Farmakokinetika lamivudinu podávaného jednou denně pediatrickým pacientům infikovaným HIV-1 ve věku od 3 měsíců do 12 let byla hodnocena ve 3 studiích (PENTA-15 [n = 17], PENTA 13 [n = 19] a ARROW PK [n = 35]). Všechny 3 studie byly 2dobé, zkřížené, otevřené farmakokinetické studie s podáváním abakaviru a lamivudinu dvakrát versus jednou denně. Tyto 3 studie prokázaly, že dávkování jednou denně poskytuje podobnou AUC0-24 jako podávání dvakrát denně lamivudinu ve stejné celkové denní dávce při porovnání dávkovacích režimů v rámci stejné formulace (tj. buď perorální roztok nebo tabletová formulace). Průměrná Cmax byla přibližně o 80 % až 90 % vyšší při dávkování lamivudinu jednou denně ve srovnání s dávkováním dvakrát denně.
Distribuce lamivudinu do mozkomíšního moku (CSF) byla hodnocena u 38 pediatrických pacientů po opakovaném perorálním podání lamivudinu. Vzorky CSF byly odebrány mezi 2 a 4 hodinami po dávce. Při dávce 8 mg na kg denně se koncentrace lamivudinu v CSF u 8 subjektů pohybovaly od 5,6 % do 30,9 % (průměr ± SD 14,2 % ± 7,9 %) koncentrace v simultánním vzorku séra, přičemž koncentrace lamivudinu v CSF se pohybovaly od 0,04 až 0,3 mcg na ml.
K dispozici jsou omezené, nekontrolované farmakokinetické a bezpečnostní údaje o podávání lamivudinu (a zidovudinu) 36 kojencům ve věku do 1 týdne ve 2 studiích v Jižní Africe. V těchto studiích byla clearance lamivudinu podstatně snížena u novorozenců ve věku 1 týdne ve srovnání s dříve studovanými pediatrickými subjekty (ve věku nad 3 měsíce). Neexistují dostatečné informace pro stanovení časového průběhu změn clearance mezi bezprostředním novorozeneckým obdobím a věkovým rozmezím starším 3 měsíců [viz NEŽÁDOUCÍ REAKCE ].
Geriatričtí pacienti
Farmakokinetika lamivudinu po podání EPIVIR 150 mg subjektům starším 65 let nebyla studována (viz Použití u konkrétních populací ].
Mužské A ženské pacienty
Ve farmakokinetice lamivudinu nejsou žádné významné nebo klinicky relevantní rozdíly mezi pohlavími.
Rasové skupiny
Ve farmakokinetice lamivudinu nejsou žádné významné nebo klinicky relevantní rasové rozdíly.
Studie lékových interakcí
Vliv lamivudinu na farmakokinetiku jiných látek
Na základě výsledků studie in vitro se neočekává, že by lamivudin při terapeutických expozicích ovlivňoval farmakokinetiku léků, které jsou substráty následujících transportérů: transportní polypeptid organického aniontu 1B1/3 (OATP1B1/3), protein rezistence rakoviny prsu (BCRP), P-glykoprotein (P-gp), vícelékový a toxinový extruzní protein 1 (MATE)1, MATE2-K, transportér organického kationtu 1 (OCT)1, OCT2 nebo OCT3.
Účinek jiných látek na farmakokinetiku lamivudinu
Lamivudin je substrátem MATE1, MATE2-K a OCT2 in vitro. Bylo prokázáno, že trimethoprim (inhibitor těchto lékových transportérů) zvyšuje plazmatické koncentrace lamivudinu. Tato interakce se nepovažuje za klinicky významnou, protože není nutná úprava dávky lamivudinu.
Lamivudin je substrátem P-gp a BCRP; vzhledem k jeho absolutní biologické dostupnosti (87 %) je však nepravděpodobné, že by tyto transportéry hrály významnou roli v absorpci lamivudinu. Proto je nepravděpodobné, že by současné podávání léků, které jsou inhibitory těchto efluxních transportérů, ovlivnilo vylučování a vylučování lamivudinu.
Interferon Alfa
Ve studii s 19 zdravými mužskými subjekty nedošlo k žádné významné farmakokinetické interakci mezi lamivudinem a interferonem alfa (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Ribavirin
Údaje in vitro naznačují, že ribavirin snižuje fosforylaci lamivudinu, stavudinu a zidovudinu. Nebyly však pozorovány žádné farmakokinetické (např. plazmatické koncentrace nebo intracelulární koncentrace trifosforylovaného aktivního metabolitu) nebo farmakodynamické (např. ztráta virologické suprese HIV-1/HCV) interakce, když ribavirin a lamivudin (n = 18), stavudin (n = 10) nebo zidovudin (n = 6) byly souběžně podávány jako součást režimu s více léky subjektům koinfikovaným HIV-1/HCV [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Sorbitol (pomocná látka)
Lamivudin a roztoky sorbitolu byly společně podávány 16 zdravým dospělým subjektům v otevřené, randomizované, 4dobé, zkřížené studii. Každý subjekt dostal jednu 300mg dávku perorálního roztoku lamivudinu samotného nebo společně s jednou dávkou 3,2 gramů, 10,2 gramů nebo 13,4 gramů sorbitolu v roztoku. Současné podávání lamivudinu se sorbitolem vedlo k poklesu AUC(0-24) závislým na dávce o 20 %, 39 % a 44 %, AUC(∞) o 14 %, 32 % a 36 % a 28 %, 52 % a 55 % v Cmax; lamivudinu, resp.
trimethoprim/sulfamethoxazol
Lamivudin a TMP/SMX byly současně podávány 14 HIV-1 pozitivním subjektům v jednocentrické, otevřené, randomizované, zkřížené studii. Každý subjekt dostával léčbu jednou dávkou 300 mg lamivudinu a TMP 160 mg/SMX 800 mg jednou denně po dobu 5 dnů se současným podáváním 300 mg lamivudinu s pátou dávkou ve zkříženém uspořádání. Současné podávání TMP/SMX s lamivudinem vedlo ke zvýšení AUC∞ lamivudinu o 43 % ± 23 % (průměr ± SD), snížení perorální clearance lamivudinu o 29 % ± 13 % a snížení o 30 % ± 36 % renální clearance lamivudinu. Farmakokinetické vlastnosti TMP a SMX nebyly při současném podávání s lamivudinem změněny. Neexistují žádné informace týkající se účinku vyšších dávek TMP/SMX, jako jsou ty, které se používají k léčbě PCP, na farmakokinetiku lamivudinu.
zidovudin
Žádné klinicky významné změny farmakokinetiky lamivudinu nebo zidovudinu nebyly pozorovány u 12 asymptomatických dospělých jedinců infikovaných HIV-1, kterým byla podána jedna dávka zidovudinu (200 mg) v kombinaci s opakovanými dávkami lamivudinu (300 mg každých 12 hodin).
Mikrobiologie
Mechanismus působení
Lamivudin je syntetický nukleosidový analog. Intracelulárně je lamivudin fosforylován na svůj aktivní 5'-trifosfátový metabolit, lamivudintrifosfát (3TC-TP). Hlavním způsobem účinku 3TC-TP je inhibice HIV-1 reverzní transkriptázy (RT) prostřednictvím ukončení řetězce DNA po začlenění nukleotidového analogu.
Antivirová aktivita
Antivirová aktivita lamivudinu proti HIV-1 byla hodnocena v řadě buněčných linií včetně monocytů a čerstvých lidských lymfocytů periferní krve (PBMC) pomocí standardních testů citlivosti. Hodnoty EC50 byly v rozmezí 0,003 až 15 mikroM (1 mikroM = 0,23 mcg na ml). Střední hodnoty EC50 lamivudinu byly 60 nM (rozsah: 20 až 70 nM), 35 nM (rozsah: 30 až 40 nM), 30 nM (rozsah: 20 až 90 nM), 20 nM (rozsah: 3 až 40 nM) 30 nM (rozsah: 1 až 60 nM), 30 nM (rozsah: 20 až 70 nM), 30 nM (rozsah: 3 až 70 nM) a 30 nM (rozsah: 20 až 90 nM) proti kladům HIV-1 AG a viry skupiny O (n = 3 kromě n = 2 pro klad B). Hodnoty EC50 proti izolátům HIV-2 (n = 4) se pohybovaly v rozmezí od 0,003 do 0,120 mikroM v PBMC. Lamivudin nebyl antagonistický ke všem testovaným anti-HIV látkám. Ribavirin (50 mikroM) používaný při léčbě chronické infekce HCV snížil anti-HIV-1 aktivitu lamivudinu 3,5krát v buňkách MT-4.
Odpor
buněčné kultuře byly selektovány varianty HIV-1 rezistentní na lamivudin. Genotypová analýza ukázala, že rezistence byla způsobena specifickou substitucí aminokyselin v reverzní transkriptáze HIV-1 v kodonu 184, která mění methionin buď na valin nebo isoleucin (M184V/I).
Od subjektů byly izolovány kmeny HIV-1 rezistentní na lamivudin i zidovudin. Citlivost klinických izolátů na lamivudin a zidovudin byla sledována v kontrolovaných klinických studiích. U subjektů užívajících lamivudin v monoterapii nebo v kombinaci s lamivudinem a zidovudinem se izoláty HIV-1 od většiny subjektů staly fenotypově a genotypově rezistentní na lamivudin během 12 týdnů.
Genotypová a fenotypová analýza izolátů HIV-1 během terapie od subjektů s virologickým selháním
Zkušební verze EPV20001
Padesát tři z 554 (10 %) subjektů zařazených do EPV20001 bylo identifikováno jako virologické selhání (hladina plazmatické HIV-1 RNA vyšší nebo rovna 400 kopiím na ml) do týdne 48. Dvacet osm subjektů bylo randomizováno do lamivudinu jednou- denně léčené skupině a 25 skupině léčené lamivudinem dvakrát denně. Střední výchozí plazmatické hladiny HIV-1 RNA subjektů ve skupině s lamivudinem jednou denně a lamivudinem dvakrát denně byly 4,9 log10 kopií na ml a 4,6 log10 kopií na ml, v daném pořadí.
Genotypová analýza izolátů při léčbě od 22 subjektů identifikovaných jako virologické selhání ve skupině s lamivudinem podávaným jednou denně ukázala, že izoláty od 8 z 22 subjektů obsahovaly substituci související s rezistencí na lamivudin (M184V nebo M184I) související s léčbou, izoláty od 0 z 22 subjekty obsahovaly substituce aminokyselin související s rezistencí na zidovudin (M41L, D67N, K70R, L210W, T215Y/F nebo K219Q/E) a izoláty od 10 z 22 subjektů obsahovaly substituce aminokyselin související s rezistencí na efavirenz ( L100I, K101E, K103N, V108I nebo Y181C).
Genotypová analýza izolátů při léčbě od subjektů (n = 22) ve skupině léčené lamivudinem dvakrát denně ukázala, že izoláty od 5 z 22 subjektů obsahovaly substituce rezistence na lamivudin vzniklé při léčbě, izoláty od 1 z 22 subjektů vykazovaly rezistenci na zidovudin vznikající při léčbě substituce a izoláty od 7 z 22 subjektů obsahovaly substituce rezistence na efavirenz vzniklé při léčbě.
Fenotypová analýza izolátů HIV-1 odpovídajících výchozímu stavu při léčbě od subjektů (n = 13), kteří dostávali lamivudin jednou denně, ukázala, že izoláty od 7 ze 13 subjektů vykazovaly 85 až 299násobné snížení citlivosti na lamivudin, izoláty od 12 13 subjektů bylo citlivých na zidovudin a izoláty od 8 ze 13 subjektů vykazovaly 25 až 295násobné snížení citlivosti na efavirenz.
Fenotypová analýza izolátů HIV-1 odpovídajících výchozímu stavu při léčbě od subjektů (n = 13), kteří dostávali lamivudin dvakrát denně, ukázala, že izoláty od 4 ze 13 subjektů vykazovaly 29 až 159násobné snížení citlivosti na lamivudin, izoláty ze všech 13 subjekty byly citlivé na zidovudin a izoláty od 3 ze 13 subjektů vykazovaly 21 až 342násobné snížení citlivosti na efavirenz.
Zkušební verze EPV40001
Padesát subjektů dostávalo lamivudin 300 mg jednou denně plus zidovudin 300 mg dvakrát denně plus abakavir 300 mg dvakrát denně a 50 subjektů dostávalo lamivudin 150 mg plus zidovudin 300 mg plus abakavir 300 mg dvakrát denně. Střední výchozí plazmatické hladiny HIV-1 RNA pro subjekty ve 2 skupinách byly 4,79 log10 kopií na ml a 4,83 log10 kopií na ml. Čtrnáct z 50 subjektů ve skupině léčené lamivudinem jednou denně a 9 z 50 subjektů ve skupině léčené lamivudinem dvakrát denně bylo identifikováno jako virologické selhání.
Genotypová analýza izolátů HIV-1 při léčbě od subjektů (n = 9) ve skupině léčené lamivudinem jednou denně ukázala, že izoláty od 6 subjektů měly samotnou substituci M184V spojenou s abakavirem a/nebo lamivudinem. Izoláty během terapie od subjektů (n = 6), kteří dostávali lamivudin dvakrát denně, prokázaly, že izoláty od 2 subjektů měly samotný M184V a izoláty od 2 subjektů obsahovaly substituci M184V v kombinaci se substitucemi aminokyselin souvisejícími s rezistencí na zidovudin.
Fenotypová analýza izolátů při léčbě od subjektů (n = 6), kteří dostávali lamivudin jednou denně, ukázala, že izoláty HIV-1 od 4 subjektů vykazovaly 32 až 53násobné snížení citlivosti na lamivudin. Izoláty HIV-1 od těchto 6 subjektů byly citlivé na zidovudin.
Fenotypová analýza izolátů při léčbě od subjektů (n = 4), kteří dostávali lamivudin dvakrát denně, ukázala, že izoláty HIV-1 od 1 subjektu vykazovaly 45násobné snížení citlivosti na lamivudin a 4,5násobné snížení citlivosti na zidovudin.
Pediatrie
Pediatričtí pacienti užívající perorální roztok lamivudinu současně s jinými antiretrovirovými perorálními roztoky (abakavir, nevirapin/efavirenz nebo zidovudin) v ARROW vyvinuli virovou rezistenci častěji než u pacientů užívajících tablety. Při randomizaci k dávkování jednou nebo dvakrát denně EPIVIR plus abakavir mělo 13 % subjektů, které začaly s tabletami, a 32 % subjektů, které začaly s roztokem, substituční rezistenci. Profil rezistence pozorovaný v pediatrii je podobný profilu pozorovanému u dospělých, pokud jde o detekované genotypové substituce a relativní frekvenci, přičemž nejčastěji zjištěné substituce jsou na M184 (V nebo I) [viz Klinické studie ].
Křížový odpor
Mezi nukleosidovými inhibitory reverzní transkriptázy (NRTI) byla pozorována zkřížená rezistence. Mutanty HIV-1 rezistentní na lamivudin byly v buněčné kultuře zkříženě rezistentní vůči didanosinu (ddl). Zkřížená rezistence se očekává také u abakaviru a emtricitabinu, protože tyto substituce M184V vybírají.
Klinické studie
Použití EPIVIR je založeno na výsledcích klinických studií u jedinců infikovaných HIV-1 v kombinovaných režimech s jinými antiretrovirovými látkami. Informace ze studií s klinickými cíli nebo kombinací počtu CD4+ buněk a měření HIV-1 RNA jsou uvedeny níže jako dokumentace příspěvku lamivudinu ke kombinovanému režimu v kontrolovaných studiích.
Dospělé subjekty
Klinická koncová zkouška
NUCB3007 (CAESAR) byla multicentrická, dvojitě zaslepená, placebem kontrolovaná studie srovnávající pokračující současnou léčbu (samotný zidovudin [62 % subjektů] nebo zidovudin s didanosinem nebo zalcitabinem [38 % subjektů]) s přidáním EPIVIR 150 mg nebo EPIVIR 150 mg plus zkoumaný nenukleosidový inhibitor reverzní transkriptázy (NNRTI), randomizováno 1:2:1. Celkem bylo zařazeno 1 816 dospělých infikovaných HIV-1 s 25 až 250 CD4+ buňkami na mm³ (medián = 122 buněk na mm³) na začátku studie: střední věk byl 36 let, 87 % byli muži, 84 % měli zkušenosti s nukleosidy a 16 % bylo terapeuticky naivních. Medián trvání studie byl 12 měsíců. Výsledky jsou shrnuty v tabulce 9.
Zkoušky náhradního koncového bodu
Duální nukleosidové analogové zkoušky
Hlavní klinické studie v počátečním vývoji lamivudinu porovnávaly kombinace lamivudin/zidovudin s monoterapií zidovudinem nebo se zidovudinem plus zalcitabin. Tyto studie prokázaly antivirový účinek lamivudinu v kombinaci 2 léčiv. Novější použití lamivudinu při léčbě infekce HIV-1 jej začleňuje do režimů s více léky obsahujícími alespoň 3 antiretrovirová léčiva pro zesílenou virovou supresi.
Srovnání dávkových režimů Testy náhradního koncového bodu u terapeuticky naivních dospělých
EPV20001 byla multicentrická, dvojitě zaslepená, kontrolovaná studie, ve které byli pacienti randomizováni v poměru 1:1, aby dostávali EPIVIR 300 mg jednou denně nebo EPIVIR 150 mg dvakrát denně v kombinaci se zidovudinem 300 mg dvakrát denně a efavirenzem 600 mg jednou denně. Celkem bylo zapsáno 554 antiretrovirovou léčbou dosud neléčených dospělých infikovaných HIV-1: muž (79 %), bílý (50 %), střední věk 35 let, výchozí počet buněk CD4+ 69 až 1 089 buněk na mm³ (medián = 362 buněk na mm³) a střední výchozí plazmatická HIV-1 RNA 4,66 log10 kopií na ml. Výsledky léčby po dobu 48 týdnů jsou shrnuty na obrázku 1 a tabulce 10.
Obrázek 1: Virologická odpověď do 48. týdne, EPV20001a,b (záměr léčby) 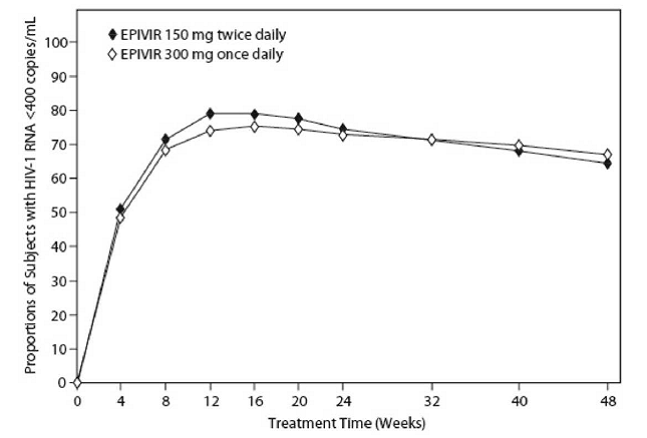
monitor Roche AMPLICOR HIV-1. b Reagujícími na každou návštěvu jsou jedinci, kteří dosáhli a udrželi HIV-1 RNA méně než 400 kopií na ml bez přerušení touto návštěvou.
Podíl subjektů s HIV-1 RNA méně než 50 kopií na ml (přes Roche Ultrasensitive test) do týdne 48 byl 61 % u subjektů užívajících EPIVIR 300 mg jednou denně a 63 % u subjektů užívajících EPIVIR 150 mg dvakrát denně. Medián zvýšení počtu CD4+ buněk byl 144 buněk na mm³ v týdnu 48 u subjektů užívajících EPIVIR 300 mg jednou denně a 146 buněk na mm³ u subjektů užívajících EPIVIR 150 mg dvakrát denně.
Malá, randomizovaná, otevřená pilotní studie EPV40001 byla provedena v Thajsku. Bylo zařazeno celkem 159 dosud neléčených dospělých subjektů (muži 32 %, Asiaté 100 %, medián věku 30 let, výchozí medián počtu CD4+ buněk 380 buněk na mm³, medián plazmatické HIV-1 RNA 4,8 log10 kopií na ml). Dvě z léčebných ramen v této studii poskytla srovnání mezi lamivudinem 300 mg jednou denně (n = 54) a lamivudinem 150 mg dvakrát denně (n = 52), každé v kombinaci se zidovudinem 300 mg dvakrát denně a abakavirem 300 mg dvakrát denně. V analýzách intent-to-treat 48týdenních dat byl podíl subjektů s HIV-1 RNA pod 400 kopií na ml 61 % (33 z 54) ve skupině randomizované na lamivudin podávaný jednou denně a 75 % (39 z 52) ve skupině randomizované k užívání všech 3 léků dvakrát denně; podíly s HIV-1 RNA pod 50 kopií na ml byly 54 % (29 z 54) ve skupině užívající lamivudin jednou denně a 67 % (35 z 52) ve skupině užívající pouze dvakrát denně; a střední zvýšení počtu CD4+ buněk bylo 166 buněk na mm³ ve skupině užívající lamivudin jednou denně a 216 buněk na mm³ ve skupině užívající dvakrát denně.
Pediatrické předměty
Klinická koncová zkouška
ACTG300 byla multicentrická, randomizovaná, dvojitě zaslepená studie, která poskytovala srovnání EPIVIR plus RETROVIR (zidovudin) s monoterapií didanosinem. Do těchto 2 léčebných ramen bylo zařazeno celkem 471 symptomatických, HIV-1-infikovaných terapií dosud neléčených (méně než nebo rovných 56 dnům antiretrovirové terapie) pediatrických pacientů. Střední věk byl 2,7 roku (rozmezí: 6 týdnů až 14 let), 58 % byly ženy a 86 % byly jiné než bílé pleti. Průměrný výchozí počet buněk CD4+ byl 868 buněk na mm³ (průměr: 1 060 buněk na mm³ a rozmezí: 0 až 4 650 buněk na mm³ pro subjekty ve věku do 5 let; průměr: 419 buněk na mm³ a rozmezí: 0 až 1 555 buněk na mm³ u subjektů starších 5 let) a průměrná výchozí plazmatická HIV-1 RNA byla 5,0 log10 kopií na ml. Medián trvání studie byl 10,1 měsíce u subjektů užívajících EPIVIR 150 mg plus RETROVIR a 9,2 měsíce u subjektů užívajících didanosin v monoterapii. Výsledky jsou shrnuty v tabulce 11.
Dávkování jednou denně
ARROW (COL105677) byla 5letá randomizovaná, multicentrická studie, která hodnotila různé aspekty klinické léčby infekce HIV-1 u pediatrických subjektů. Pacienti infikovaní HIV-1 dosud neléčení ve věku od 3 měsíců do 17 let byli zařazeni a léčeni režimem první linie obsahujícím EPIVIR 150 mg a abakavir v dávce dvakrát denně podle doporučení Světové zdravotnické organizace. Po minimálně 36 týdnech léčby byla subjektům dána možnost zúčastnit se Randomizace 3 studie ARROW, porovnávající bezpečnost a účinnost dávkování jednou denně s dávkováním EPIVIR a abakaviru dvakrát denně v kombinaci s třetím antiretrovirovým léku po dobu dalších 96 týdnů. Z 1 206 původních subjektů ARROW se 669 zúčastnilo Randomizace 3. Virologická suprese nebyla podmínkou účasti: na začátku Randomizace 3 (po minimálně 36 týdnech léčby dvakrát denně) 75 % subjektů při léčbě dvakrát denně kohorty byly virologicky suprimovány ve srovnání se 71 % subjektů v kohortě užívající jednou denně.
Podíl subjektů s HIV-1 RNA méně než 80 kopií na ml během 96 týdnů je uveden v tabulce 12. Rozdíly mezi virologickými odpověďmi ve dvou léčebných ramenech byly srovnatelné napříč základními charakteristikami pro pohlaví a věk.
Analýzy podle formulace prokázaly podíl subjektů s HIV-1 RNA méně než 80 kopií na ml při randomizaci a v týdnu 96 byl vyšší u subjektů, které dostávaly tablety EPIVIR 150 mg a abakavir (75 % [458/610] a 72 % [434/601]) než u těch, kteří dostávali roztok(y) (s roztokem EPIVIR 150 mg podávaným v dávkách založených na hmotnostním pásmu přibližně 8 mg na kg za den) kdykoli (52 % [29/56] a 54 % [30/56]), respektive [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]. Tyto rozdíly byly pozorovány u každé různé hodnocené věkové skupiny.
INFORMACE PRO PACIENTA
EPIVIR (EP-i-veer) (lamivudin) tablety
EPIVIR (EP-i-veer) (lamivudin) perorální roztok
Jaké jsou nejdůležitější informace, které bych měl vědět o přípravku EPIVIR 150 mg?
EPIVIR může způsobit závažné nežádoucí účinky, včetně:
- Zhoršení viru hepatitidy B u lidí, kteří mají infekci HIV-1. Pokud máte infekci HIV-1 (virus lidské imunodeficience typu 1) a virovou hepatitidu B (HBV), může se vaše HBV zhoršit (vzplanutí), pokud přestanete přípravek EPIVIR užívat. „Vzplanutí“ je, když se vaše infekce HBV náhle vrátí horším způsobem než dříve. Zhoršení onemocnění jater může být vážné a může vést ke smrti.
- Nenechte si dojít EPIVIR. Doplňte si předpis nebo si promluvte se svým poskytovatelem zdravotní péče, než bude váš EPIVIR celý pryč.
- Neukončujte léčbu přípravkem EPIVIR, aniž byste si nejprve promluvili se svým poskytovatelem zdravotní péče.
- Pokud přestanete užívat přípravek EPIVIR, váš poskytovatel zdravotní péče bude muset často kontrolovat váš zdravotní stav a pravidelně po dobu několika měsíců provádět krevní testy, aby zkontroloval vaše játra.
- Rezistentní virus hepatitidy B (HBV). Pokud máte HIV-1 a hepatitidu B, může se virus hepatitidy B během léčby přípravkem EPIVIR 150 mg změnit (mutovat) a stát se hůře léčitelným (odolným).
- Použití s režimy na bázi interferonu a ribavirinu. Ke zhoršení onemocnění jater, které způsobilo smrt, došlo u lidí infikovaných jak HIV-1, tak virem hepatitidy C, kteří užívají antiretrovirová léčiva a jsou také léčeni na hepatitidu C interferonem s ribavirinem nebo bez ribavirinu. Pokud užíváte EPIVIR 150 mg a interferon s ribavirinem nebo bez něj, informujte svého poskytovatele zdravotní péče, pokud máte nějaké nové příznaky.
Co je EPIVIR?
EPIVIR je lék na předpis používaný spolu s dalšími antiretrovirovými léky k léčbě infekce virem lidské imunodeficience (HIV-1).
HIV-1 je virus, který způsobuje syndrom získaného selhání imunity (AIDS).
Tablety a perorální roztok EPIVIR (používané k léčbě infekce HIV-1) obsahují vyšší dávku stejné účinné látky (lamivudin), než je tomu v léku EPIVIR-HBV tablety a perorální roztok (používaný k léčbě HBV). Pokud máte HIV-1 i HBV, neměli byste přípravek EPIVIR-HBV používat k léčbě infekcí.
Bezpečnost a účinnost přípravku EPIVIR u dětí mladších 3 měsíců nebyla stanovena.
Kdo by neměl užívat EPIVIR 150 mg?
Neužívejte EPIVIR jestliže jste alergický(á) na lamivudin nebo na kteroukoli další složku přípravku EPIVIR. Úplný seznam složek přípravku EPIVIR naleznete na konci této příbalové informace pro pacienty.
Co mám sdělit svému poskytovateli zdravotní péče před užitím přípravku EPIVIR?
Před užitím přípravku EPIVIR 150 mg informujte svého poskytovatele zdravotní péče, pokud:
- máte nebo jste měl(a) problémy s játry, včetně infekce virem hepatitidy B nebo C.
- mít problémy s ledvinami.
- mít cukrovku. Jedna 15ml dávka (150 mg) perorálního roztoku EPIVIR obsahuje 3 gramy sacharózy.
- jste těhotná nebo plánujete otěhotnět. Užívání přípravku EPIVIR během těhotenství nebylo spojeno se zvýšeným rizikem vrozených vad. Poraďte se se svým poskytovatelem zdravotní péče, pokud jste těhotná nebo plánujete otěhotnět. Registr těhotenství. Pro ženy, které během těhotenství užívají antiretrovirová léčiva, existuje těhotenský registr. Účelem tohoto registru je shromažďovat informace o zdraví vás a vašeho dítěte. Promluvte si se svým poskytovatelem zdravotní péče o tom, jak se můžete zúčastnit tohoto registru.
- kojíte nebo plánujete kojit. Pokud užíváte EPIVIR, nekojte.
- Pokud máte HIV-1, neměla byste kojit kvůli riziku přenosu HIV-1 na vaše dítě.
Informujte svého poskytovatele zdravotní péče o všech lécích, které užíváte, včetně léků na předpis a volně prodejných léků, vitamínů a bylinných doplňků.
Některé léky interagují s EPIVIRem. Uchovávejte si seznam svých léků a ukažte jej svému poskytovateli zdravotní péče a lékárníkovi, když dostanete nový lék. Můžete požádat svého poskytovatele zdravotní péče nebo lékárníka o seznam léků, které interagují s přípravkem EPIVIR.
Nezačínejte užívat nový lék, aniž byste o tom informovali svého poskytovatele zdravotní péče. Váš poskytovatel zdravotní péče vám může říci, zda je bezpečné užívat EPIVIR s jinými léky.
Jak mám užívat EPIVIR?
- Užívejte EPIVIR přesně tak, jak vám řekl váš poskytovatel zdravotní péče.
- Pokud vynecháte dávku přípravku EPIVIR 150 mg, vezměte si ji, jakmile si vzpomenete. Neužívejte 2 dávky současně ani neužívejte více, než vám lékař řekl, abyste si vzali.
- Během léčby přípravkem EPIVIR zůstaňte v péči poskytovatele zdravotní péče.
- EPIVIR lze užívat s jídlem nebo bez jídla.
- Dětem ve věku 3 měsíců a starším lékař předepíše dávku přípravku EPIVIR 150 mg na základě tělesné hmotnosti vašeho dítěte.
- Informujte svého poskytovatele zdravotní péče, pokud máte vy nebo vaše dítě potíže s polykáním tablet. EPIVIR 150 mg se dodává také jako tekutina (perorální roztok).
- Nenechte si dojít EPIVIR. Virus ve vaší krvi se může zvýšit a virus se může hůře léčit. Když vám začne docházet zásoba, získejte další od svého poskytovatele zdravotní péče nebo v lékárně.
- Pokud užijete příliš mnoho přípravku EPIVIR 150 mg, zavolejte svého poskytovatele zdravotní péče nebo jděte ihned na pohotovost v nejbližší nemocnici.
Jaké jsou možné nežádoucí účinky přípravku EPIVIR 150 mg?
- EPIVIR 150 mg může způsobit závažné nežádoucí účinky včetně:
- Viz "Jaké jsou nejdůležitější informace, které bych měl vědět o přípravku EPIVIR 150 mg?"
- Tvorba kyseliny v krvi (laktátová acidóza). U některých lidí, kteří užívají EPIVIR, se může objevit laktátová acidóza. Laktátová acidóza je vážná lékařská pohotovost, která může způsobit smrt. Okamžitě zavolejte svého poskytovatele zdravotní péče, pokud se u Vás objeví některý z následujících příznaků, které by mohly být příznaky laktátové acidózy:
- cítit se velmi slabý nebo unavený
- cítit chlad, zejména v pažích a nohou
- neobvyklá (ne normální) bolest svalů
- cítit závrať nebo točení hlavy
- potíže s dýcháním
- máte rychlý nebo nepravidelný srdeční tep
- bolest žaludku s nevolností a zvracením
- Vážné problémy s játry se může stát u lidí, kteří užívají EPIVIR. V některých případech mohou tyto závažné jaterní problémy vést ke smrti. Vaše játra se mohou zvětšit (hepatomegalie) a může se u vás vytvořit tuk v játrech (steatóza). Okamžitě zavolejte svého poskytovatele zdravotní péče, pokud se u Vás objeví některý z následujících příznaků nebo příznaků jaterních problémů:
- vaše kůže nebo bílá část vašich očí zežloutne (žloutenka)
- ztráta chuti k jídlu na několik dní nebo déle
- nevolnost
- tmavá nebo „čajově zbarvená“ moč
- bolest, bolest nebo citlivost na pravé straně oblasti žaludku
- světlá stolice (pohyby střev)
Pokud jste žena nebo máte velkou nadváhu (obézní), může být pravděpodobnější, že dostanete laktátovou acidózu nebo závažné problémy s játry.
- Riziko zánětu slinivky břišní (pankreatitida). Děti mohou být ohroženy rozvojem pankreatitidy během léčby přípravkem EPIVIR, pokud:
- užívali léky na bázi nukleosidových analogů
- měli v minulosti pankreatitidu
- mají jiné rizikové faktory pro pankreatitidu
Okamžitě zavolejte svého poskytovatele zdravotní péče, pokud se u vašeho dítěte objeví známky a příznaky pankreatitidy včetně silné bolesti v horní části žaludku, s nevolností a zvracením nebo bez nich. Váš poskytovatel zdravotní péče vám může říci, abyste přestali podávat EPIVIR 150 mg svému dítěti, pokud jeho příznaky a výsledky krevních testů ukazují, že vaše dítě může mít pankreatitidu.
- Změny ve vašem imunitním systému (imunitní rekonstituční syndrom) se může stát, když začnete užívat léky proti HIV-1. Váš imunitní systém může zesílit a začít bojovat s infekcemi, které byly ve vašem těle ukryty dlouhou dobu. Okamžitě informujte svého poskytovatele zdravotní péče, pokud začnete mít nové příznaky poté, co začnete užívat EPIVIR.
Mezi nejčastější nežádoucí účinky přípravku EPIVIR 150 mg u dospělých patří:
- bolest hlavy
- nosní příznaky a symptomy
- nevolnost
- průjem
- obecně se necítí dobře
- únava
- kašel
Mezi nejčastější nežádoucí účinky přípravku EPIVIR 150 mg u dětí patří horečka a kašel.
Informujte svého poskytovatele zdravotní péče, pokud máte jakýkoli vedlejší účinek, který vás obtěžuje nebo který nezmizí.
Toto nejsou všechny možné vedlejší účinky přípravku EPIVIR. Zavolejte svého lékaře o radu ohledně nežádoucích účinků. Nežádoucí účinky můžete hlásit úřadu FDA na čísle 1-800-FDA-1088.
Jak mám EPIVIR uchovávat?
- Uchovávejte EPIVIR 150 mg tablety a perorální roztok při pokojové teplotě mezi 68 °F až 77 °F (20 °C až 25 °C).
- Lahvičky s perorálním roztokem EPIVIR 150 mg udržujte těsně uzavřené.
Uchovávejte EPIVIR a všechny léky mimo dosah dětí.
Obecné informace o bezpečném a účinném používání přípravku EPIVIR.
Léky jsou někdy předepisovány pro jiné účely, než které jsou uvedeny v příbalové informaci pro pacienty. Nepoužívejte EPIVIR na stav, pro který nebyl předepsán. Nepodávejte EPIVIR jiným lidem, i když mají stejné příznaky jako vy. Může jim to ublížit.
Můžete požádat svého poskytovatele zdravotní péče nebo lékárníka o informace o přípravku EPIVIR 150 mg, který je určen pro zdravotníky.
Pro více informací navštivte www.viivhealthcare.com nebo volejte 1-877-844-8872.
Jaké jsou složky přípravku EPIVIR 150 mg?
Účinná látka: lamivudin
Neaktivní složky:
EPIVIR 150 mg 150 mg potahované tablety: hypromelóza, magnesium-stearát, mikrokrystalická celulóza, polyethylenglykol, polysorbát 80, sodná sůl glykolátu škrobu a oxid titaničitý.
EPIVIR 300 mg potahované tablety: černý oxid železitý, hypromelóza, magnesium-stearát, mikrokrystalická celulóza, polyethylenglykol, polysorbát 80, sodná sůl glykolátu škrobu a oxid titaničitý.
EPIVIR 150 mg perorální roztok: umělé jahodové a banánové aroma, kyselina citronová (bezvodá), methylparaben, propylenglykol, propylparaben, citrát sodný (dihydrát) a sacharóza (200 mg na ml).
Tyto informace pro pacienty byly schváleny americkým Úřadem pro kontrolu potravin a léčiv.