Rebetol 200mg Ribavirin Použití, vedlejší účinky a dávkování. Cena v internetové lékárně. Generické léky bez předpisu.
Co je Rebetol 200 mg a jak se používá?
Rebetol je lék na předpis používaný k léčbě příznaků chronické hepatitidy C. Rebetol 200 mg lze užívat samostatně nebo s jinými léky.
Rebetol patří do třídy léků nazývaných hepatitidy B/hepatitidy C; Agenti RSV.
Není známo, zda je přípravek Rebetol 200 mg bezpečný a účinný u dětí mladších 3 let.
Jaké jsou možné vedlejší účinky přípravku Rebetol?
Rebetol může způsobit závažné nežádoucí účinky, včetně:
- kopřivka,
- potíže s dýcháním,
- otok obličeje, rtů, jazyka nebo hrdla,
- problémy se zrakem
- silná bolest v horní části žaludku šířící se do zad,
- nevolnost,
- zvracení,
- průjem,
- nový nebo zhoršující se kašel,
- horečka,
- bodavá bolest na hrudi,
- sípání,
- dušnost,
- těžká deprese,
- myšlenky na sebepoškozování,
- myšlenky na ubližování druhým,
- bledá nebo zažloutlá kůže,
- tmavě zbarvená moč,
- zmatek,
- slabost,
- zimnice,
- příznaky podobné chřipce,
- oteklé dásně,
- vředy v ústech,
- kožní vředy,
- snadná tvorba modřin,
- neobvyklé krvácení a
- točení hlavy
- Pokud máte některý z výše uvedených příznaků, okamžitě vyhledejte lékařskou pomoc.
- Mezi nejčastější vedlejší účinky přípravku Rebetol patří:
- nevolnost,
- zvracení,
- ztráta chuti k jídlu,
- horečka,
- zimnice,
- třes,
- nízký počet krvinek,
- anémie,
- slabost,
- únava,
- bolest hlavy,
- bolest svalů,
- změny nálady,
- úzkost a
- podrážděnost
Informujte lékaře, pokud máte jakýkoli nežádoucí účinek, který vás obtěžuje nebo který neustupuje.
To nejsou všechny možné vedlejší účinky přípravku Rebetol. Pro více informací se zeptejte svého lékaře nebo lékárníka.
Zavolejte svého lékaře o radu ohledně nežádoucích účinků. Nežádoucí účinky můžete hlásit úřadu FDA na čísle 1-800-FDA-1088.
VAROVÁNÍ
RIZIKO VÁŽNÝCH PORUCH A ÚČINKŮ SPOJENÝCH S RIBAVIRINEM
- Monoterapie REBETOL není účinná při léčbě chronické infekce virem hepatitidy C a neměla by být pro tuto indikaci používána samostatně (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ).
- Primární toxicitou ribavirinu je hemolytická anémie. Anémie spojená s léčbou REBETOL 200 mg může vést ke zhoršení srdečního onemocnění, které vedlo k fatálním a nefatálním infarktům myokardu. Pacienti s anamnézou významného nebo nestabilního srdečního onemocnění by neměli být léčeni přípravkem REBETOL (viz DÁVKOVÁNÍ A PODÁVÁNÍ, VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ a NEŽÁDOUCÍ ÚČINKY ).
- všech druhů zvířat vystavených ribavirinu byly prokázány významné teratogenní a embryocidní účinky. Kromě toho má ribavirin poločas vícenásobných dávek 12 dní, a tak může přetrvávat v neplazmatických kompartmentech po dobu až 6 měsíců. Proto je terapie REBETOLem kontraindikována u těhotných žen a u mužských partnerů těhotných žen. Extrémní opatrnosti je třeba dbát na to, aby se zabránilo otěhotnění během léčby a 6 měsíců po jejím ukončení jak u pacientek, tak u partnerek mužů, kteří užívají léčbu REBETOLem. Během léčby a během 6měsíčního období sledování po léčbě musí být použity alespoň dvě spolehlivé formy účinné antikoncepce (viz KONTRAINDIKACE, VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ, Použití u specifických populací, Neklinická toxikologie a INFORMACE PRO PACIENTA ).
POPIS
REBETOL (ribavirin) je syntetický nukleosidový analog (purinový analog). Chemický název ribavirinu je 1-β-D-ribofuranosyl-1H-1,2,4-triazol-3-karboxamid a má následující strukturní vzorec (viz obrázek 1).
Obrázek 1: Strukturní vzorec
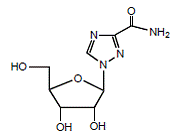
Ribavirin je bílý krystalický prášek. Je volně rozpustný ve vodě a mírně rozpustný v bezvodém alkoholu. Empirický vzorec je C8H12N4O5 a molekulová hmotnost je 244,21.
REBETOL tobolky se skládají z bílého prášku v bílé, neprůhledné, želatinové tobolce. Každá tobolka obsahuje 200 mg ribavirinu a neaktivní složky mikrokrystalickou celulózu, monohydrát laktózy, sodnou sůl kroskarmelózy a magnesium-stearát. Obal kapsle se skládá z želatiny, laurylsulfátu sodného, oxidu křemičitého a oxidu titaničitého. Kapsle je potištěna jedlým modrým farmaceutickým inkoustem, který je vyroben ze šelaku, bezvodého ethylalkoholu, isopropylalkoholu, n-butylalkoholu, propylenglykolu, hydroxidu amonného a hliníkového laku FD&C Blue #2.
REBETOL 200 mg perorální roztok je čirá, bezbarvá až světle nebo světle žlutá tekutina s příchutí žvýkačky. Každý mililitr roztoku obsahuje 40 mg ribavirinu a neaktivní složky sacharózu, glycerin, sorbitol, propylenglykol, citrát sodný, kyselinu citrónovou, benzoát sodný, přírodní a umělé aroma pro žvýkačku #15864 a vodu.
INDIKACE
Chronická hepatitida C (CHC)
REBETOL® (ribavirin) v kombinaci s interferonem alfa-2b (pegylovaným a nepegylovaným) je indikován k léčbě chronické hepatitidy C (CHC) u pacientů ve věku 3 let a starších s kompenzovaným onemocněním jater [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ , a Použití ve specifických populacích ].
Při zahájení kombinované terapie REBETOL s PegIntron® nebo INTRON A® je třeba vzít v úvahu následující body:
- Tyto indikace jsou založeny na dosažení nedetekovatelné HCV-RNA po léčbě po dobu 24 nebo 48 týdnů a udržení trvalé virologické odpovědi (SVR) 24 týdnů po poslední dávce.
- Kombinovaná terapie s REBETOL/PegIntron je preferována před REBETOL/INTRON A, protože tato kombinace poskytuje podstatně lepší míru odezvy [viz Klinické studie ].
- pacientů s následujícími charakteristikami je méně pravděpodobné, že budou mít prospěch z opětovné léčby poté, co selhal léčebný cyklus: předchozí nereakce, předchozí léčba pegylovaným interferonem, významná přemosťující fibróza nebo cirhóza a infekce genotypu 1 [viz Klinické studie ].
- Pro léčbu delší než jeden rok nejsou k dispozici žádné údaje o bezpečnosti a účinnosti.
DÁVKOVÁNÍ A PODÁVÁNÍ
Kapsle REBETOL se za žádných okolností nesmí otevírat, drtit nebo lámat. REBETOL 200 mg se má užívat s jídlem [viz KLINICKÁ FARMAKOLOGIE ]. REBETOL by neměl být používán u pacientů s clearance kreatininu nižší než 50 ml/min.
Kombinovaná terapie REBETOL/PegIntron
Dospělí pacienti
Doporučená dávka PegIntronu je 1,5 mcg/kg/týden subkutánně v kombinaci s 800 až 1400 mg REBETOL 200 mg tobolky perorálně na základě tělesné hmotnosti pacienta (viz tabulka 1). Objem PegIntronu, který má být injikován, závisí na síle PegIntronu a tělesné hmotnosti pacienta, další informace o dávkování viz označení PegIntronu.
Délka léčby – pacienti dosud neléčení interferonem alfa
Délka léčby u pacientů s genotypem 1 je 48 týdnů. Přerušení léčby by mělo být zváženo u pacientů, kteří nedosáhnou poklesu nebo ztráty HCV-RNA alespoň o 2 log10 ve 12. týdnu, nebo pokud HCV-RNA zůstává detekovatelná po 24 týdnech léčby. Pacienti s genotypem 2 a 3 by měli být léčeni po dobu 24 týdnů.
Délka léčby – Opakovaná léčba pomocí PegIntron/REBETOL v případě selhání předchozí léčby
Délka léčby u pacientů, u kterých dříve selhala terapie, je 48 týdnů, bez ohledu na genotyp HCV. Přeléčení pacienti, kteří nedosáhnou nedetekovatelné HCV-RNA ve 12. týdnu terapie nebo jejichž HCV-RNA zůstává detekovatelná po 24 týdnech terapie, je vysoce nepravděpodobné, že dosáhnou SVR a je třeba zvážit přerušení léčby [viz Klinické studie ].
Pediatričtí pacienti
Dávkování u pediatrických pacientů se u PegIntronu určuje podle tělesného povrchu au REBETOLu podle tělesné hmotnosti. Doporučená dávka PegIntronu je 60 mcg/m²/týden subkutánně v kombinaci s 15 mg/kg/den přípravku REBETOL 200 mg perorálně ve dvou dílčích dávkách (viz tabulka 2) pro pediatrické pacienty ve věku 3-17 let. Pacienti, kteří během léčby přípravkem PegIntron/REBETOL 200 mg dosáhnou 18. narozenin, by měli zůstat v pediatrickém dávkovacím režimu. Délka léčby u pacientů s genotypem 1 je 48 týdnů. Pacienti s genotypem 2 a 3 by měli být léčeni po dobu 24 týdnů.
REBETOL/INTRON A Kombinovaná terapie
Dospělí
Délka léčby – pacienti dosud neléčení interferonem alfa
Doporučená dávka přípravku INTRON A je 3 miliony IU třikrát týdně subkutánně. Doporučená dávka tobolek REBETOL závisí na tělesné hmotnosti pacienta (viz tabulka 3). Doporučená délka léčby u pacientů, kteří dříve neléčili interferonem, je 24 až 48 týdnů. Délka léčby by měla být individuální pro pacienta v závislosti na výchozích charakteristikách onemocnění, odpovědi na léčbu a snášenlivosti režimu [viz INDIKACE A POUŽITÍ , NEŽÁDOUCÍ REAKCE , a Klinické studie ]. Po 24 týdnech léčby by měla být vyhodnocena virologická odpověď. U každého pacienta, který nedosáhl HCV-RNA pod limitem detekce testu do 24 týdnů, je třeba zvážit přerušení léčby. Neexistují žádné údaje o bezpečnosti a účinnosti léčby trvající déle než 48 týdnů u dříve neléčené populace pacientů.
Délka léčby – Opakovaná léčba přípravkem INTRON A/REBETOL u pacientů s relapsem
pacientů s relapsem po monoterapii nepegylovaným interferonem je doporučená délka léčby 24 týdnů.
Pediatrie
Doporučená dávka přípravku REBETOL je 15 mg/kg denně perorálně (rozdělená dávka AM a PM). Dávkování přípravku REBETOL pro děti v kombinaci s přípravkem INTRON A naleznete v tabulce 2. INTRON A pro injekci podle tělesné hmotnosti 25 kg až 61 kg je 3 miliony IU/m² třikrát týdně subkutánně. Pro více než 61 kg tělesné hmotnosti viz tabulka dávkování pro dospělé.
Doporučená délka léčby je 48 týdnů u pediatrických pacientů s genotypem 1. Po 24 týdnech léčby by měla být vyhodnocena virologická odpověď. Přerušení léčby by mělo být zváženo u každého pacienta, který do této doby nedosáhl HCV-RNA pod limitem detekce testu. Doporučená délka léčby u pediatrických pacientů s genotypem 2/3 je 24 týdnů.
Laboratorní testy
Následující laboratorní testy se doporučují všem pacientům léčeným přípravkem REBETOL 200 mg před zahájením léčby a poté pravidelně po ní.
Standardní hematologické testy – včetně hemoglobinu (předběžná léčba, týden 2 a týden 4 léčby a podle klinicky vhodného [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ], kompletní a diferenciální počet bílých krvinek a počet krevních destiček.
- Chemie krve - jaterní testy a TSH.
- Těhotenství – včetně měsíčního sledování u žen ve fertilním věku.
- EKG [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Modifikace dávky
Pokud se během kombinované terapie REBETOL/INTRON A nebo terapie REBETOL/PegIntron vyvinou závažné nežádoucí reakce nebo laboratorní abnormality, upravte nebo přerušte dávku, dokud nežádoucí reakce nezmizí nebo nesníží závažnost (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]. Pokud intolerance přetrvává i po úpravě dávky, kombinovaná léčba by měla být přerušena. Snížení dávky PegIntronu u dospělých pacientů na kombinované terapii REBETOL/PegIntron se provádí ve dvou krocích z původní počáteční dávky 1,5 mcg/kg/týden na 1 mcg/kg/týden a poté na 0,5 mcg/kg/týden , V případě potřeby. Další informace týkající se snížení dávky PegIntronu naleznete na štítku PegIntronu.
Ve studii 2 kombinované terapie pro dospělé došlo ke snížení dávky u 42 % subjektů užívajících PegIntron 1,5 mcg/kg a REBETOL 800 mg denně, včetně 57 % subjektů s hmotností 60 kg nebo méně. Ve studii 4 došlo u 16 % subjektů ke snížení dávky PegIntronu na 1 mcg/kg v kombinaci s REBETOLem, přičemž další 4 % vyžadovala druhé snížení dávky PegIntronu na 0,5 mcg/kg kvůli nežádoucím účinkům (viz NEŽÁDOUCÍ REAKCE ].
Snížení dávky u pediatrických pacientů se provádí úpravou doporučené dávky PegIntronu ve dvou krocích z původní počáteční dávky 60 mcg/m²/týden na 40 mcg/m²/týden a poté na 20 mcg/m²/týden, pokud potřebné (viz tabulka 4). Ve studii pediatrické kombinované terapie došlo ke snížení dávky u 25 % subjektů užívajících PegIntron 60 mcg/m² týdně a REBETOL 15 mg/kg denně. Snížení dávky u pediatrických pacientů je dosaženo úpravou doporučené dávky REBETOLu z původní počáteční dávky 15 mg/kg denně ve dvou krocích na 12 mg/kg/den a poté na 8 mg/kg/den, pokud je to nutné ( viz tabulka 4).
REBETOL 200 mg by se neměl používat u pacientů s clearance kreatininu nižší než 50 ml/min. Pacienti s poruchou funkce ledvin a osoby starší 50 let by měly být pečlivě sledovány s ohledem na rozvoj anémie (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ , Použití u konkrétních populací , a KLINICKÁ FARMAKOLOGIE ].
REBETOL 200 mg by měl být podáván s opatrností pacientům s již existujícím srdečním onemocněním. Pacienti by měli být vyšetřeni před zahájením léčby a během léčby by měli být náležitě sledováni. Pokud dojde ke zhoršení kardiovaskulárního stavu, léčba by měla být ukončena [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
U pacientů se stabilním kardiovaskulárním onemocněním v anamnéze je nutné trvalé snížení dávky, pokud hemoglobin poklesne o více než nebo rovný 2 g/dl během kteréhokoli 4týdenního období. Navíc u těchto pacientů se srdeční anamnézou, pokud hemoglobin zůstane nižší než 12 g/dl po 4 týdnech při snížené dávce, pacient by měl přerušit kombinovanou léčbu.
Doporučuje se, aby pacientovi, jehož hladina hemoglobinu klesne pod 10 g/dl, byla dávka REBETOLu upravena nebo vysazena podle tabulky 4 [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Další informace o tom, jak snížit dávku přípravku INTRON A nebo PegIntron, naleznete na štítku pro INTRON A nebo PegIntron.
Přerušení dávkování
Dospělí
pacientů s HCV genotypu 1, u pacientů dosud neléčených interferonem alfa, kteří dostávají PegIntron v kombinaci s ribavirinem, se doporučuje přerušení léčby, pokud nedojde k poklesu nebo ztrátě HCV-RNA alespoň o 2 log10 po 12 týdnech léčby nebo pokud HCV-RNA hladiny zůstávají detekovatelné po 24 týdnech terapie. Bez ohledu na genotyp je u dříve léčených pacientů, kteří mají detekovatelnou HCV-RNA ve 12. nebo 24. týdnu vysoce nepravděpodobné, že dosáhnou SVR, a je třeba zvážit přerušení léčby.
Pediatrie (3–17 let)
Doporučuje se, aby pacienti užívající kombinaci PegIntron/REBETOL (kromě HCV genotypu 2 a 3) přerušili léčbu po 12 týdnech, pokud jejich léčba 12. týden HCV-RNA poklesla o méně než 2 log10 ve srovnání s předléčbou, nebo po 24 týdnech, pokud mají detekovatelné HCV-RNA při léčbě 24. týden.
JAK DODÁVÁNO
Dávkové formy A Síly
REBETOL 200 mg kapsle 200 mg
REBETOL 200 mg perorální roztok 40 mg na ml
Skladování A Manipulace
REBETOL 200 mg tobolky jsou bílé, neprůhledné tobolky s REBETOL, 200 mg, a logem Schering Corporation vytištěným na obalu tobolky; tobolky jsou baleny v lahvičce obsahující 56 tobolek ( NDC 0085-1351-05), 70 kapslí ( NDC 0085-1385-07) a 84 kapslí ( NDC 0085-1194-03).
REBETOL 200 mg perorální roztok 40 mg na ml je čirá, bezbarvá až bledě nebo světle žlutá kapalina s příchutí žvýkačky a je balena v 4 oz jantarových skleněných lahvičkách (100 ml/láhev) s uzávěry odolnými proti otevření dětmi ( NDC 0085-1318-01).
Lahvička tobolek REBETOL by měla být skladována při 25°C (77°F); povoleny výchylky do 15-30°C (59-86°F) [viz USP řízená pokojová teplota ].
REBETOL 200 mg perorální roztok by měl být skladován při teplotě 2-8°C (36-46°F) nebo při 25°C (77°F); povoleny výchylky do 15-30°C (59-86°F) [viz USP řízená pokojová teplota ].
REBETOL 200 mg perorální roztok vyrobený pro: Merck Sharp & Dohme Corp., dceřiná společnost Whitehouse Station, NJ 08889, USA. Výrobce: Schering-Plough Canada, Inc., Pointe Claire, Quebec, Kanada Kapsle REBETOL vyrábí: Merck Sharp & Dohme Corp., dceřiná společnost Whitehouse Station, NJ 08889, USA. Revize: prosinec 2014.
VEDLEJŠÍ EFEKTY
Klinické studie s REBETOLem v kombinaci s PegIntronem nebo INTRONem A byly provedeny u více než 7800 subjektů ve věku od 3 do 76 let.
Primární toxicitou ribavirinu je hemolytická anémie. K poklesu hladin hemoglobinu došlo během prvních 1 až 2 týdnů perorální léčby. Srdeční a plicní reakce spojené s anémií se vyskytly přibližně u 10 % pacientů [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Více než 96 % všech subjektů v klinických studiích zaznamenalo jednu nebo více nežádoucích reakcí. Nejčastěji hlášenými nežádoucími účinky u dospělých subjektů užívajících PegIntron nebo INTRON A v kombinaci s REBETOLem 200 mg byly zánět/reakce v místě vpichu, únava/astenie, bolest hlavy, zimnice, horečky, nauzea, myalgie a úzkost/emocionální labilita/podrážděnost. Nejčastějšími nežádoucími účinky u pediatrických subjektů ve věku 3 let a starších, kteří dostávali REBETOL 200 mg v kombinaci s PegIntronem nebo INTRONem A, byly pyrexie, bolest hlavy, neutropenie, únava, anorexie, erytém v místě vpichu a zvracení.
Část Nežádoucí účinky odkazuje na následující klinické studie:
- Zkoušky kombinované terapie REBETOL/PegIntron:
- Klinická studie 1 – hodnocená monoterapie PegIntronem (v tomto štítku není dále popsána; informace o této studii viz označení PegIntronu).
- Studie 2 – hodnotila REBETOL 800 mg/den paušální dávku v kombinaci s 1,5 mcg/kg/týden PegIntronem nebo s INTRONem A.
- Studie 3 – hodnotila PegIntron/režim REBETOL na základě hmotnosti 200 mg v kombinaci s režimem PegIntron/jednotková dávka REBETOL 200 mg.
- Studie 4 – porovnávala dvě dávky PegIntronu (1,5 mcg/kg/týden a 1 mcg/kg/týden) v kombinaci s REBETOLem a třetí léčebnou skupinu, která dostávala Pegasys® (180 mcg/týden)/Copegus® (1000-1200 mg/den) ).
- Studie 5 – hodnotila PegIntron (1,5 mcg/kg/týden) v kombinaci s přípravkem REBETOL založeným na hmotnosti u jedinců, kteří dříve selhali při léčbě.
- PegIntron/REBETOL 200 mg kombinovaná terapie u dětských pacientů
- REBETOL/INTRON A Testy kombinované terapie pro dospělé a pediatry
Závažné nežádoucí účinky se objevily u přibližně 12 % subjektů v klinických studiích s PegIntronem s REBETOLem nebo bez něj (viz VAROVÁNÍ V KRABICE , VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]. Nejčastějšími závažnými příhodami vyskytujícími se u subjektů léčených přípravkem PegIntron a REBETOL 200 mg byly deprese a sebevražedné myšlenky (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ], každý se vyskytuje s frekvencí menší než 1 %. Sebevražedné myšlenky nebo pokusy se vyskytovaly častěji u pediatrických pacientů, především dospívajících, ve srovnání s dospělými pacienty (2,4 % oproti 1 %) během léčby a sledování mimo terapii (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]. Nejčastější fatální reakcí vyskytující se u subjektů léčených PegIntronem a REBETOL 200 mg byla zástava srdce, sebevražedné myšlenky a pokusy o sebevraždu (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ], všechny se vyskytují u méně než 1 % subjektů.
Vzhledem k tomu, že klinické studie jsou prováděny za velmi odlišných podmínek, nelze míry nežádoucích reakcí pozorované v klinických studiích léku přímo srovnávat s mírami v klinických studiích jiného léku a nemusí odrážet míry pozorované v klinické praxi.
Zkušenosti z klinických studií – kombinovaná terapie REBETOL/PegIntron
Dospělé subjekty
Nežádoucí účinky, které se vyskytly v klinické studii s incidencí vyšší než 5 %, jsou uvedeny u léčebné skupiny z kombinované terapie REBETOL/PegIntron (studie 2) v tabulce 5.
Tabulka 6 shrnuje nežádoucí reakce související s léčbou ve studii 4, které se vyskytly s incidencí vyšší nebo rovnou 10 %.
Výskyt závažných nežádoucích účinků byl ve všech studiích srovnatelný. Ve studii 3 byla podobná incidence závažných nežádoucích účinků hlášena u skupiny REBETOL na základě hmotnosti (12 %) a v režimu s plochými dávkami REBETOL. Ve studii 2 byla incidence závažných nežádoucích účinků 17 % ve skupinách užívajících PegIntron/REBETOL ve srovnání se 14 % ve skupině INTRON A/REBETOL.
mnoha, ale ne ve všech případech, nežádoucí účinky vymizely po snížení dávky nebo přerušení léčby. U některých subjektů se během 6měsíčního období sledování objevily přetrvávající nebo nové závažné nežádoucí reakce. Ve studii 2 mnoho subjektů nadále pociťovalo nežádoucí reakce několik měsíců po přerušení léčby. Na konci 6měsíčního období sledování byla incidence probíhajících nežádoucích účinků podle tělesné třídy ve skupině PegIntron 1,5/REBETOL 200 mg 33 % (psychiatrická), 20 % (muskuloskeletální) a 10 % (pro endokrinní a pro GI). U přibližně 10 až 15 % subjektů se úbytek hmotnosti, únava a bolest hlavy nevyřešily.
těchto klinických studiích došlo během léčby nebo během sledování k úmrtí 31 subjektů. Ve studii 1 došlo k 1 sebevraždě u subjektu léčeného PegIntronem v monoterapii a ke 2 úmrtím u subjektů užívajících monoterapii INTRONem A (1 vražda/sebevražda a 1 náhlá smrt). Ve studii 2 došlo k 1 sebevraždě u subjektu, který dostával kombinovanou terapii PegIntron/REBETOL 200 mg; a úmrtí 1 subjektu ve skupině INTRON A/REBETOL 200 mg (nehoda motorového vozidla). Ve studii 3 došlo ke 14 úmrtím, z nichž 2 byly pravděpodobné sebevraždy a 1 nevysvětlené úmrtí u osoby s příslušnou anamnézou deprese. Ve studii 4 došlo k 12 úmrtím, z nichž 6 se vyskytlo u jedinců, kteří dostávali kombinovanou terapii PegIntron/REBETOL, 5 ve skupině PegIntron 1,5 mcg/REBETOL (N=1019) a 1 ve skupině PegIntron 1 mcg/REBETOL (N= 1016) a 6 z nich se vyskytlo u subjektů užívajících Pegasys/Copegus (N=1035); došlo ke 3 sebevraždám, ke kterým došlo během období sledování mimo léčbu u subjektů, které dostávaly kombinovanou terapii PegIntron (1,5 mcg/kg)/REBETOL 200 mg.
Ve studiích 1 a 2 přerušilo léčbu 10 až 14 % subjektů užívajících PegIntron samotný nebo v kombinaci s REBETOLem 200 mg ve srovnání s 6 % léčenými samotným INTRONem A a 13 % léčenými INTRONem A v kombinaci s REBETOLem. Podobně ve studii 3 15 % subjektů užívajících PegIntron v kombinaci s REBETOLem podle hmotnosti a 14 % subjektů užívajících PegIntron a paušální dávku REBETOL 200 mg přerušilo léčbu kvůli nežádoucí reakci. Nejčastější důvody pro přerušení léčby souvisely se známými účinky interferonu na psychiatrické, systémové (např. únava, bolest hlavy) nebo gastrointestinální nežádoucí účinky. Ve studii 4 13 % subjektů ve skupině PegIntron 1,5 mcg/REBETOL 200 mg, 10 % ve skupině PegIntron 1 mcg/REBETOL 200 mg a 13 % ve skupině Pegasys 180 mcg/Copegus přerušilo léčbu kvůli nežádoucím účinkům.
Ve studii 2 se snížení dávky v důsledku nežádoucích účinků objevilo u 42 % subjektů užívajících PegIntron (1,5 mcg/kg)/REBETOL 200 mg a u 34 % pacientů užívajících INTRON A/REBETOL. Většina subjektů (57 %) s hmotností 60 kg nebo méně, kterým byl podáván PegIntron (1,5 mcg/kg)/REBETOL, vyžadovala snížení dávky. Snížení interferonu bylo závislé na dávce (PegIntron 1,5 mcg/kg vyšší než PegIntron 0,5 mcg/kg nebo INTRON A), 40 %, 27 %, 28 %, v tomto pořadí. Snížení dávky REBETOL 200 mg bylo podobné ve všech třech skupinách, 33 až 35 %. Nejčastějšími důvody pro úpravu dávky byla neutropenie (18 %) nebo anémie (9 %) (viz Laboratorní hodnoty ). Mezi další časté důvody patřila deprese, únava, nevolnost a trombocytopenie. Ve studii 3 se úpravy dávky v důsledku nežádoucích účinků vyskytovaly častěji u dávkování na základě hmotnosti (WBD) ve srovnání s plochým dávkováním (29 %, resp. 23 %). Ve studii 4 došlo u 16 % subjektů ke snížení dávky PegIntronu na 1 mcg/kg v kombinaci s REBETOL 200 mg, přičemž další 4 % vyžadovala druhé snížení dávky PegIntronu na 0,5 mcg/kg v důsledku nežádoucích účinků ve srovnání s 15 % pacientů v rameni Pegasys/Copegus, kteří vyžadovali snížení dávky na 135 mcg/týden u Pegasysu, s dalšími 7 % ve skupině Pegasys/Copegus vyžadující druhé snížení dávky na 90 mcg/týden u Pegasysu.
Ve studiích s kombinací PegIntron/REBETOL 200 mg byly nejčastějšími nežádoucími účinky psychiatrické, které se vyskytly u 77 % subjektů ve studii 2 a u 68 % až 69 % subjektů ve studii 3. Tyto psychiatrické nežádoucí účinky zahrnovaly nejčastěji depresi, podrážděnost a nespavost, každou hlášenou přibližně 30 % až 40 % subjektů ve všech léčebných skupinách. K sebevražednému chování (myšlenkám, pokusům a sebevraždám) došlo u 2 % všech subjektů během léčby nebo během sledování po ukončení léčby (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]. Ve studii 4 se psychiatrické nežádoucí účinky vyskytly u 58 % subjektů v rameni PegIntron 1,5 mcg/REBETOL 200 mg, 55 % subjektů v rameni PegIntron 1 mcg/REBETOL 200 mg a 57 % subjektů v rameni Pegasyscg/Copem 180 .
PegIntron vyvolal únavu nebo bolest hlavy u přibližně dvou třetin subjektů, s horečkou nebo zimnicí u přibližně poloviny subjektů. Závažnost některých z těchto systémových příznaků (např. horečka a bolest hlavy) měla tendenci klesat, jak léčba pokračovala. Ve studiích 1 a 2 se zánět a reakce v místě aplikace (např. modřina, svědění a podráždění) objevily přibližně dvakrát častěji u terapií PegIntron (až u 75 % subjektů) ve srovnání s přípravkem INTRON A. Bolest v místě vpichu však byla vzácné (2 až 3 %) ve všech skupinách. Ve studii 3 byl celkový výskyt reakcí v místě vpichu nebo zánětu 23 % až 24 %.
Subjekty, které dostávaly REBETOL/PegIntron jako opakovanou léčbu po selhání předchozího kombinovaného režimu s interferonem, hlásily během klinických studií dosud neléčených subjektů nežádoucí účinky podobné těm, které byly dříve spojeny s tímto režimem.
Pediatrické předměty
Obecně byl profil nežádoucích účinků u pediatrické populace podobný jako u dospělých. V pediatrické studii byly nejčastějšími nežádoucími účinky u všech subjektů pyrexie (80 %), bolest hlavy (62 %), neutropenie (33 %), únava (30 %), anorexie (29 %), erytém v místě vpichu (29 %) a zvracení (27 %). Většina nežádoucích účinků hlášených ve studii byla mírná nebo středně závažná. Závažné nežádoucí účinky byly hlášeny u 7 % (8/107) všech subjektů a zahrnovaly bolest v místě vpichu (1 %), bolest končetin (1 %), bolest hlavy (1 %), neutropenii (1 %) a pyrexii (4 %). Důležité nežádoucí reakce, které se vyskytly u této populace, byly nervozita (7 %; 7/107), agresivita (3 %; 3/107), hněv (2 %; 2/107) a deprese (1 %; 1/107). . Pět subjektů dostalo léčbu levothyroxinem, tři s klinickou hypotyreózou a dva s asymptomatickým zvýšením TSH. Přírůstek hmotnosti a výšky u pediatrických subjektů léčených přípravkem PegIntron plus REBETOL zaostával za tím, co předpovídaly údaje o normativní populaci po celou dobu léčby. Silně inhibovaná rychlost růstu (méně než 3. percentil) byla pozorována u 70 % subjektů během léčby.
Úprava dávky PegIntronu a/nebo ribavirinu byla nutná u 25 % subjektů v důsledku nežádoucích účinků souvisejících s léčbou, nejčastěji pro anémii, neutropenii a úbytek hmotnosti. Dva jedinci (2 %; 2/107) přerušili léčbu v důsledku nežádoucí reakce.
Nežádoucí účinky, které se vyskytly s incidencí vyšší nebo rovnou 10 % u subjektů pediatrické studie, jsou uvedeny v tabulce 7.
Devadesát čtyři ze 107 subjektů bylo zařazeno do pětileté dlouhodobé následné studie. Dlouhodobé účinky na růst byly menší u subjektů léčených po dobu 24 týdnů než u subjektů léčených po dobu 48 týdnů. U 24 procent subjektů (11/46) léčených po dobu 24 týdnů a u 40 % subjektů (19/48) léčených po dobu 48 týdnů došlo k > 15 percentilovému snížení výšky vzhledem k věku od doby před léčbou do konce 5 let dlouhodobé sledování ve srovnání se základními percentily před léčbou. U 11 procent subjektů (5/46) léčených po dobu 24 týdnů a u 13 % subjektů (6/48) léčených po dobu 48 týdnů bylo pozorováno snížení od výchozí hodnoty před léčbou o > 30 percentilů výšky vzhledem k věku do konce z 5letého dlouhodobého sledování. Přestože bylo pozorováno napříč všemi věkovými skupinami, zdálo se, že nejvyšší riziko snížení výšky na konci dlouhodobého sledování koreluje se zahájením kombinované terapie během let očekávané maximální rychlosti růstu. [Vidět VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]
Laboratorní hodnoty
Dospělí a dětští pacienti
Profil nežádoucích účinků ve studii 3, která srovnávala kombinaci PegIntron/REBETOL 200 mg na základě hmotnosti s režimem PegIntron/jednotková dávka REBETOL 200 mg, odhalil zvýšený výskyt anémie s dávkováním podle hmotnosti (29 % vs. 19 % u dávkování založeného na hmotnosti vs. režimy s plochými dávkami). Většina případů anémie však byla mírná a reagovala na snížení dávky.
Změny vybraných laboratorních hodnot během léčby v kombinaci s léčbou REBETOL 200mg jsou popsány níže. Snížení hemoglobinu, leukocytů, neutrofilů a krevních destiček může vyžadovat snížení dávky nebo trvalé ukončení léčby [vidět DÁVKOVÁNÍ A PODÁVÁNÍ ]. Změny vybraných laboratorních hodnot během terapie jsou popsány v tabulce 8. Většina změn laboratorních hodnot ve studii PegIntron/REBETOL 200 mg s pediatry byla mírná nebo středně závažná.
Hemoglobin
Hladiny hemoglobinu se snížily na méně než 11 g/dl u přibližně 30 % subjektů ve studii 2. Ve studii 3 mělo 47 % subjektů užívajících WBD REBETOL 200 mg a 33 % na ploché dávce REBETOL 200 mg pokles hladin hemoglobinu nižší než 11 g /dl. Snížení hemoglobinu na méně než 9 g/dl se objevilo častěji u subjektů užívajících WBD ve srovnání s plochým dávkováním (4 % a 2 %). Ve studii 2 byla nutná úprava dávky u 9 % a 13 % subjektů ve skupinách PegIntron/REBETOL 200 mg a INTRON A/REBETOL 200 mg. Ve studii 4 měly subjekty užívající PegIntron (1,5 mcg/kg)/REBETOL pokles hladin hemoglobinu na 8,5 až méně než 10 g/dl (28 %) a na méně než 8,5 g/dl (3 %), zatímco u pacientů užívající Pegasys 180 mcg/Copegus k tomuto poklesu došlo u 26 % au 4 % subjektů. Hladiny hemoglobinu se stabilizovaly v průměru ve 4. až 6. týdnu léčby. Typickým pozorovaným vzorem byl pokles hladin hemoglobinu v léčebném týdnu 4, po kterém následovala stabilizace a plató, které se udrželo až do konce léčby. V monoterapeutické studii PegIntron byly poklesy hemoglobinu obecně mírné a úpravy dávky byly zřídka nutné (viz DÁVKOVÁNÍ A PODÁVÁNÍ ].
Neutrofily
Snížení počtu neutrofilů bylo pozorováno u většiny dospělých subjektů léčených kombinovanou terapií s REBETOL 200 mg ve studii 2 (85 %) a INTRON A/REBETOL (60 %). Závažná potenciálně život ohrožující neutropenie (méně než 0,5 x 109/l) se objevila u 2 % subjektů léčených přípravkem INTRON A/REBETOL 200 mg a přibližně u 4 % subjektů léčených přípravkem PegIntron/REBETOL 200 mg ve studii 2. Osmnáct procent subjektů užívajících PegIntron/REBETOL ve studii 2 vyžadoval úpravu dávkování interferonu. Několik subjektů (méně než 1 %) vyžadovalo trvalé přerušení léčby. Počty neutrofilů se obecně vrátily na úroveň před léčbou 4 týdny po ukončení léčby [viz DÁVKOVÁNÍ A PODÁVÁNÍ ].
Krevní destičky
Počet krevních destiček se snížil na méně než 100 000/mm³ u přibližně 20 % subjektů léčených samotným PegIntronem nebo REBETOLem 200 mg au 6 % dospělých subjektů léčených přípravkem INTRON A/REBETOL. K závažnému poklesu počtu krevních destiček (méně než 50 000/mm³) dochází u méně než 4 % dospělých subjektů. Pacienti mohou vyžadovat přerušení nebo úpravu dávky v důsledku poklesu krevních destiček [viz DÁVKOVÁNÍ A PODÁVÁNÍ ]. Ve studii 2 vyžadovalo 1 % nebo 3 % subjektů úpravu dávky INTRONu A nebo PegIntronu, v daném pořadí. Počet krevních destiček se obecně vrátil na úroveň před léčbou 4 týdny po ukončení léčby.
Funkce štítné žlázy
Vývoj abnormalit TSH, s klinickými projevy nebo bez nich, je spojen s terapií interferony. Ve studii 2 se klinicky zjevné poruchy štítné žlázy vyskytly u subjektů léčených buď INTRONem A nebo PegIntronem (s REBETOLem nebo bez něj) s podobnou incidencí (5 % pro hypotyreózu a 3 % pro hypertyreózu). U subjektů se objevily nové abnormality TSH během léčby a během období sledování. Na konci období sledování mělo 7 % subjektů stále abnormální hodnoty TSH.
Bilirubin a kyselina močová
Ve studii 2 se u 10 až 14 % subjektů rozvinula hyperbilirubinémie au 33 až 38 % hyperurikemie v souvislosti s hemolýzou. U šesti subjektů se vyvinula mírná až středně závažná dna.
Zkušenosti z klinických studií – REBETOL/INTRON A kombinovaná terapie
Dospělé subjekty
klinických studiích 19 % a 6 % dříve neléčených subjektů a 6 % subjektů s relapsem, v daném pořadí, přerušilo léčbu kvůli nežádoucím účinkům v kombinovaných ramenech ve srovnání s 13 % a 3 % v ramenech s interferonem. Vybrané nežádoucí reakce související s léčbou, které se vyskytly ve studiích v USA s incidencí vyšší nebo rovnou 5 %, jsou uvedeny podle léčebné skupiny (viz tabulka 9). Obecně byly vybrané nežádoucí účinky související s léčbou hlášeny s nižší incidencí v mezinárodních studiích ve srovnání se studiemi v USA, s výjimkou astenie, symptomů podobných chřipce, nervozity a pruritu.
Pediatrické předměty
klinických studiích u 118 pediatrických pacientů ve věku 3 až 16 let 6 % přerušilo léčbu z důvodu nežádoucích účinků. Úprava dávky byla nutná u 30 % subjektů, nejčastěji pro anémii a neutropenii. Obecně byl profil nežádoucích účinků u pediatrické populace podobný jako u dospělých. Poruchy v místě vpichu, horečka, anorexie, zvracení a emoční labilita se vyskytovaly častěji u pediatrických subjektů ve srovnání s dospělými subjekty. Naopak dětští jedinci pociťovali menší únavu, dyspepsii, artralgii, nespavost, podrážděnost, poruchu koncentrace, dušnost a svědění ve srovnání s dospělými jedinci. Vybrané nežádoucí účinky související s léčbou, které se vyskytly s incidencí vyšší nebo rovnou 5 % u všech pediatrických pacientů, kteří dostávali doporučenou dávku kombinované terapie REBETOL/INTRON A, jsou uvedeny v tabulce 9.
Během 48týdenní léčebné kúry došlo ke snížení rychlosti lineárního růstu (průměrné snížení percentilového přiřazení o 7 %) a snížení rychlosti nárůstu hmotnosti (průměrné snížení percentilového přiřazení o 9 %). Obecný obrat těchto trendů byl zaznamenán během 24týdenního období po léčbě. Dlouhodobé údaje u omezeného počtu pacientů však naznačují, že kombinovaná léčba může vyvolat inhibici růstu, která u některých pacientů vede ke snížení konečné výšky v dospělosti (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Laboratorní hodnoty
Změny vybraných hematologických hodnot (hemoglobin, bílé krvinky, neutrofily a krevní destičky) během terapie jsou popsány níže (viz tabulka 10).
Hemoglobin. Pokles hemoglobinu u subjektů léčených REBETOLem začal v týdnu 1, se stabilizací do týdne 4. U dříve neléčených subjektů léčených po dobu 48 týdnů byl průměrný maximální pokles od výchozí hodnoty 3,1 g/dl v americké studii a 2,9 g/dl v mezinárodní studii. soud. U pacientů s relapsem bylo průměrné maximální snížení od výchozí hodnoty 2,8 g/dl v americké studii a 2,6 g/dl v mezinárodní studii. Hodnoty hemoglobinu se u většiny subjektů vrátily na úroveň před léčbou během 4 až 8 týdnů po ukončení terapie.
Bilirubin a kyselina močová. V klinických studiích bylo zaznamenáno zvýšení bilirubinu i kyseliny močové spojené s hemolýzou. Většinou se jednalo o středně závažné biochemické změny, které byly zvráceny do 4 týdnů po ukončení léčby. Toto pozorování se vyskytovalo nejčastěji u subjektů s předchozí diagnózou Gilbertova syndromu. To nebylo spojeno s jaterní dysfunkcí nebo klinickou morbiditou.
Postmarketingové zkušenosti
Následující nežádoucí účinky byly identifikovány a hlášeny během používání přípravku REBETOL po schválení v kombinaci s přípravkem INTRON A nebo PegIntron. Protože jsou tyto reakce hlášeny dobrovolně z populace nejisté velikosti, není vždy možné spolehlivě odhadnout jejich frekvenci nebo stanovit příčinnou souvislost s expozicí léku.
Poruchy krve a lymfatického systému
Čistá aplazie červených krvinek, aplastická anémie
Poruchy ucha a labyrintu
Porucha sluchu, vertigo
Respirační, hrudní a mediastinální poruchy
Plicní Hypertenze
Oční poruchy
Serózní odchlípení sítnice
Endokrinní poruchy
Diabetes
DROGOVÉ INTERAKCE
didanosin
Expozice didanosinu nebo jeho aktivnímu metabolitu (dideoxyadenosin 5'-trifosfát) se zvyšuje, pokud je didanosin podáván společně s ribavirinem, což může způsobit nebo zhoršit klinickou toxicitu; proto je současné podávání REBETOL 200 mg tobolky nebo perorálního roztoku a didanosinu kontraindikováno. V klinických studiích byly hlášeny případy fatálního selhání jater, stejně jako periferní neuropatie, pankreatitida a symptomatická hyperlaktatémie/laktátová acidóza.
Analogy nukleosidů
pacientů s cirhózou koinfikovaných HIV/HCV, kteří dostávali kombinovanou antiretrovirovou léčbu HIV a interferon alfa a ribavirin, se objevila jaterní dekompenzace (některá fatální). Přidání léčby interferony alfa samotnými nebo v kombinaci s ribavirinem může u této populace pacientů zvýšit riziko. U pacientů užívajících interferon s ribavirinem a nukleosidovými inhibitory reverzní transkriptázy (NRTI) je třeba pečlivě sledovat toxicitu související s léčbou, zejména jaterní dekompenzaci a anémii. Vysazení NRTI by mělo být považováno za lékařsky vhodné (viz označení pro jednotlivé produkty NRTI ). Pokud je pozorována zhoršující se klinická toxicita, včetně jaterní dekompenzace (např. Child-Pugh vyšší než 6), je třeba zvážit snížení dávky nebo vysazení interferonu, ribavirinu nebo obou.
Ribavirin může antagonizovat antivirovou aktivitu stavudinu a zidovudinu na buněčné kultuře proti HIV. Bylo prokázáno, že ribavirin v buněčné kultuře inhibuje fosforylaci lamivudinu, stavudinu a zidovudinu, což by mohlo vést ke snížení antiretrovirové aktivity. Ve studii s jiným pegylovaným interferonem v kombinaci s ribavirinem však nebyly pozorovány žádné farmakokinetické (např. plazmatické koncentrace nebo koncentrace intracelulárního trifosforylovaného aktivního metabolitu) ani farmakodynamické (např. ztráta virologické suprese HIV/HCV) interakce, když ribavirin a lamivudin (n =18), stavudin (n=10) nebo zidovudin (n=6) byly souběžně podávány jako součást vícelékového režimu u subjektů koinfikovaných HIV/HCV. Současné užívání ribavirinu s některým z těchto léků by proto mělo být používáno s opatrností.
Léky metabolizované cytochromem P-450
Výsledky studií in vitro s použitím preparátů mikrosomů jater člověka i potkana ukázaly malý nebo žádný metabolismus ribavirinu zprostředkovaný enzymem cytochromu P-450 s minimálním potenciálem lékových interakcí na bázi enzymu P-450.
Ve farmakokinetické studii s více dávkami nebyly zaznamenány žádné farmakokinetické interakce mezi INTRONem A a REBETOL 200 mg tobolkami.
azathioprin
Bylo hlášeno, že použití ribavirinu k léčbě chronické hepatitidy C u pacientů užívajících azathioprin vyvolává těžkou pancytopenii a může zvýšit riziko myelotoxicity související s azathioprinem. Inosinmonofosfátdehydrogenáza (IMDH) je nezbytná pro jednu z metabolických drah azathioprinu. Je známo, že ribavirin inhibuje IMDH, což vede k akumulaci metabolitu azathioprinu, 6-methylthioinosinmonofosfátu (6-MTITP), který je spojen s myelotoxicitou (neutropenie, trombocytopenie a anémie). Pacienti, kteří dostávají azathioprin s ribavirinem, by měli mít kompletní krevní obraz, včetně počtu krevních destiček, monitorovaný první měsíc týdně, druhý a třetí měsíc léčby dvakrát měsíčně a poté měsíčně nebo častěji, pokud jsou nutné změny dávkování nebo jiné změny léčby (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
VAROVÁNÍ
Zahrnuto jako součást OPATŘENÍ sekce.
OPATŘENÍ
Těhotenství
REBETOL 200 mg tobolky a perorální roztok mohou způsobit vrozené vady a smrt nenarozeného dítěte. Léčba přípravkem REBETOL by neměla být zahájena, dokud nebude bezprostředně před plánovaným zahájením léčby získán negativní těhotenský test. Pacientky by měly během léčby a po dobu 6 měsíců po jejím ukončení používat alespoň dvě formy antikoncepce a každý měsíc by měly podstupovat těhotenské testy. Je třeba věnovat mimořádnou pozornost tomu, aby nedošlo k otěhotnění u pacientek a partnerek mužských pacientů. REBETOL 200mg se prokázal významné teratogenní a embryocidní účinky u všech druhů zvířat, u kterých byly provedeny odpovídající studie. Tyto účinky se objevily při dávkách jako nízké jako jedna dvacetina doporučené lidské dávky ribavirinu. Léčba přípravkem REBETOL 200 mg by neměla být zahájena, dokud nebude hlášen negativní těhotenský test byl získán bezprostředně před plánovaným zahájením léčby [viz VAROVÁNÍ V KRABICE , KONTRAINDIKACE , Použití u konkrétních populací , a INFORMACE PRO PACIENTA ].
Anémie
Primární toxicitou ribavirinu je hemolytická anémie, která byla v klinických studiích pozorována u přibližně 10 % subjektů léčených přípravkem REBETOL/INTRON A. Anémie spojená s tobolkami REBETOL se objeví během 1 až 2 týdnů od zahájení léčby. Protože počáteční pokles hemoglobinu může být významný, doporučuje se získat hemoglobin nebo hematokrit před zahájením léčby a ve 2. a 4. týdnu léčby nebo častěji, pokud je to klinicky indikováno. Pacienti by pak měli být sledováni, jak je to klinicky vhodné [viz DÁVKOVÁNÍ A PODÁVÁNÍ ].
pacientů s anémií způsobenou přípravkem REBETOL byly hlášeny fatální a nefatální infarkty myokardu. Před zahájením léčby ribavirinem by měli být pacienti vyšetřeni na základní srdeční onemocnění. Pacientům s již existujícím srdečním onemocněním by měly být před léčbou provedeny elektrokardiogramy a během léčby by měli být náležitě sledováni. Pokud dojde ke zhoršení kardiovaskulárního stavu, léčba by měla být pozastavena nebo přerušena [viz DÁVKOVÁNÍ A PODÁVÁNÍ ]. Vzhledem k tomu, že anémie vyvolaná léky může zhoršit srdeční onemocnění, pacienti s anamnézou významného nebo nestabilního srdečního onemocnění by neměli REBETOL používat.
Pankreatitida
Léčba REBETOL a INTRON A nebo PegIntron by měla být pozastavena u pacientů se známkami a příznaky pankreatitidy a přerušena u pacientů s potvrzenou pankreatitidou.
Plicní poruchy
Během léčby přípravkem REBETOL s kombinovanou terapií interferonem alfa byly hlášeny plicní symptomy, včetně dušnosti, plicních infiltrátů, pneumonitidy, plicní hypertenze a pneumonie; občas se vyskytly případy smrtelné pneumonie. Kromě toho byla hlášena sarkoidóza nebo exacerbace sarkoidózy. Pokud existují známky plicních infiltrátů nebo poškození plicních funkcí, pacient by měl být pečlivě sledován, a pokud je to vhodné, kombinovaná léčba by měla být přerušena.
Oftalmologické poruchy
Ribavirin se používá v kombinované terapii s interferony alfa. Snížení nebo ztráta zraku, retinopatie včetně makulárního edému, retinální tepny nebo žíly, trombóza, retinální krvácení a skvrny z vaty, optická neuritida, edém papily a serózní odchlípení sítnice jsou indukovány nebo zhoršeny léčbou interferony alfa. Všichni pacienti by měli na začátku podstoupit oční vyšetření. Pacienti s již existujícími oftalmologickými poruchami (např. diabetická nebo hypertenzní retinopatie) by měli během kombinované léčby s interferonem alfa podstupovat pravidelné oftalmologické vyšetření. Každý pacient, u kterého se rozvinou oční příznaky, by měl podstoupit rychlé a úplné oční vyšetření. Kombinovaná léčba s interferony alfa by měla být přerušena u pacientů, u kterých se rozvinou nové nebo se zhorší oftalmologické poruchy.
Laboratorní testy
PegIntron v kombinaci s ribavirinem může způsobit závažné snížení počtu neutrofilů a krevních destiček a hematologické, endokrinní (např. TSH) a jaterní abnormality.
Pacienti na kombinační léčbě PegIntron/REBETOL by měli podstoupit hematologické a biochemické vyšetření krve před zahájením léčby a poté pravidelně po ní. V klinické studii pro dospělé byl měřen kompletní krevní obraz (včetně hemoglobinu, počtu neutrofilů a krevních destiček) a chemie (včetně AST, ALT, bilirubinu a kyseliny močové) během léčebného období v týdnech 2, 4, 8, 12 a poté v 6týdenních intervalech nebo častěji, pokud se objeví abnormality. U pediatrických subjektů byly v 6. týdnu léčby hodnoceny stejné laboratorní parametry s dalším hodnocením hemoglobinu. Hladiny TSH byly měřeny každých 12 týdnů během období léčby. HCV-RNA by měla být během léčby pravidelně měřena [viz DÁVKOVÁNÍ A PODÁVÁNÍ ].
Zubní a parodontální poruchy
pacientů užívajících kombinovanou léčbu ribavirinem a interferonem nebo peginterferonem byly hlášeny poruchy zubů a parodontu. Navíc sucho v ústech může mít škodlivý účinek na zuby a sliznice úst při dlouhodobé léčbě kombinací REBETOL a pegylovaného nebo nepegylovaného interferonu alfa-2b. Pacienti by si měli dvakrát denně důkladně čistit zuby a pravidelně docházet na zubní prohlídky. Pokud dojde ke zvracení, je třeba jim doporučit, aby si poté důkladně vypláchli ústa.
Současné podávání azathioprinu
Pancytopenie (výrazný pokles počtu červených krvinek, neutrofilů a krevních destiček) a suprese kostní dřeně se v literatuře objevily během 3 až 7 týdnů po současném podání pegylovaného interferonu/ribavirinu a azathioprinu. U tohoto omezeného počtu pacientů (n=8) byla myelotoxicita reverzibilní během 4 až 6 týdnů po vysazení jak antivirové léčby HCV, tak souběžně podávaného azathioprinu a neopakovala se ani po opětovném zahájení samotné léčby. PegIntron, REBETOL a azathioprin by měly být přerušeny pro pancytopenii a pegylovaný interferon/ribavirin by neměl být znovu zaveden se současným azathioprinem (viz DROGOVÉ INTERAKCE ].
Vliv na růst - Pediatrické použití
Údaje o účincích přípravků PegIntron a REBETOL na růst pocházejí z otevřené studie u subjektů ve věku 3 až 17 let, ve které jsou změny hmotnosti a výšky srovnávány s údaji o normativní populaci v USA. Obecně platí, že přírůstek hmotnosti a výšky u pediatrických subjektů léčených PegIntronem a REBETOLem 200 mg zaostává za předpokládaným údaji o normativní populaci po celou dobu léčby. Silně inhibovaná rychlost růstu (méně než 3. percentil) byla pozorována u 70 % subjektů během léčby. Po léčbě došlo u většiny subjektů k opětovnému růstu a nárůstu hmotnosti. Údaje z dlouhodobého sledování u pediatrických pacientů však naznačují, že PegIntron v kombinované léčbě s REBETOLem může vyvolat inhibici růstu, která u některých pacientů vede ke snížení výšky v dospělosti (viz NEŽÁDOUCÍ REAKCE ].
Podobně dopad na růst byl pozorován u subjektů po léčbě přípravkem REBETOL 200 mg a kombinovanou terapií INTRON A po dobu jednoho roku. V dlouhodobé následné studii omezeného počtu těchto subjektů vedla kombinovaná terapie u některých subjektů ke snížení konečné výšky v dospělosti [viz NEŽÁDOUCÍ REAKCE ].
Záruky používání
Na základě výsledků klinických studií není monoterapie ribavirinem účinná v léčbě chronické infekce virem hepatitidy C; proto se REBETOL 200 mg tobolky nebo perorální roztok nesmí užívat samostatně. Bezpečnost a účinnost REBETOL tobolek a perorálního roztoku byla stanovena pouze při použití společně s INTRONem A nebo PegIntronem (nikoli jinými interferony) jako kombinovaná terapie.
Bezpečnost a účinnost terapie REBETOL/INTRON A a PegIntron pro léčbu infekce HIV, adenoviru, RSV, parainfluenzy nebo chřipkových infekcí nebyla stanovena. REBETOL 200 mg tobolky by neměly být používány pro tyto indikace. Ribavirin pro inhalaci má samostatné označení, které je třeba konzultovat, pokud se zvažuje inhalační léčba ribavirinem.
Existují významné nežádoucí reakce způsobené terapií REBETOL/INTRON A nebo PegIntron, včetně těžké deprese a sebevražedných myšlenek, hemolytické anémie, suprese funkce kostní dřeně, autoimunitních a infekčních poruch, plicní dysfunkce, pankreatitidy a diabetu. Sebevražedné myšlenky nebo pokusy se vyskytovaly častěji u pediatrických pacientů, především dospívajících, ve srovnání s dospělými pacienty (2,4 % oproti 1 %) během léčby a sledování mimo terapii. Označení pro INTRON A a PegIntron by mělo být před zahájením kombinované léčby zkontrolováno jako celek pro další bezpečnostní informace.
Informace pro pacienty
Doporučte pacientovi, aby si přečetl označení pacienta schválené FDA ( Průvodce léky ).
Anémie
Nejčastějším nežádoucím účinkem vyskytujícím se u tobolek REBETOL je anémie, která může být závažná (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ a NEŽÁDOUCÍ REAKCE ]. Pacienti by měli být upozorněni, že před zahájením léčby a pravidelně poté jsou vyžadována laboratorní vyšetření [viz DÁVKOVÁNÍ A PODÁVÁNÍ ]. Doporučuje se, aby pacienti byli dobře hydratováni, zejména během počátečních fází léčby.
Těhotenství
Pacienti musí být informováni, že REBETOL 200 mg tobolky a perorální roztok mohou způsobit vrozené vady a smrt nenarozeného dítěte. REBETOL 200 mg nesmí užívat těhotné ženy ani muži, jejichž partnerky jsou těhotné. Je třeba dbát mimořádné opatrnosti, aby nedošlo k otěhotnění u pacientek a partnerek mužských pacientů užívajících REBETOL. REBETOL by neměl být zahájen, dokud nebude bezprostředně před zahájením léčby získán negativní těhotenský test. Pacientky musí provádět těhotenský test měsíčně během léčby a 6 měsíců po léčbě.
Ženy ve fertilním věku musí být před zahájením léčby poučeny o používání účinné antikoncepce (dvě spolehlivé formy). Pacienti (muži a ženy) musí být informováni o teratogenních/embryocidních rizicích a musí být poučeni o používání účinné antikoncepce během REBETOL 200 mg a 6 měsíců po léčbě. Pacienti (muži a ženy) by měli být poučeni, aby v případě těhotenství okamžitě informovali lékaře [viz KONTRAINDIKACE , VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ , a Použití ve specifických populacích ].
Pokud během léčby nebo 6 měsíců po léčbě dojde k otěhotnění, musí být pacientka informována o teratogenním riziku léčby přípravkem REBETOL 200 mg pro plod. Pacientky nebo partnerky pacientů by měly svému lékaři okamžitě hlásit jakékoli těhotenství, ke kterému dojde během léčby nebo do 6 měsíců po jejím ukončení. Předepisující lékaři by měli takové případy hlásit na telefonním čísle 1-800-593-2214.
Rizika versus výhody
Pacienti užívající REBETOL tobolky by měli být informováni o přínosech a rizicích spojených s léčbou, měli by být nasměrováni na její vhodné použití a pacienti by měli být doporučeni Průvodce léky . Pacienti by měli být informováni, že účinek léčby infekce hepatitidy C na přenos není znám a že je třeba přijmout vhodná opatření k zabránění přenosu viru hepatitidy C.
Pacienti by měli být informováni o tom, co mají dělat v případě, že vynechají dávku REBETOLu; vynechanou dávku je třeba užít co nejdříve během téhož dne. Pacienti by neměli zdvojnásobit další dávku. Pacientům by mělo být doporučeno, aby se v případě dotazů obrátili na svého poskytovatele zdravotní péče.
Neklinická toxikologie
Karcinogeneze, mutageneze, zhoršení plodnosti
Karcinogeneze
Ribavirin nezpůsobil zvýšení žádného typu nádoru, když byl podáván po dobu 6 měsíců u modelu transgenních myší s deficitem p53 v dávkách až 300 mg/kg (odhadovaný lidský ekvivalent 25 mg/kg na základě úpravy tělesného povrchu pro dospělého 60 kg hmotnosti přibližně 1,9násobek maximální doporučené denní dávky pro člověka). Ribavirin byl nekarcinogenní, když byl podáván potkanům po dobu 2 let v dávkách až 40 mg/kg (odhadovaný ekvivalent u člověka 5,71 mg/kg na základě úpravy tělesného povrchu pro dospělého o hmotnosti 60 kg).
Mutageneze
Ribavirin prokázal zvýšený výskyt mutací a buněčné transformace v četných testech genotoxicity. Ribavirin byl aktivní v testu Balb/3T3 in vitro buněčné transformace. Mutagenní aktivita byla pozorována při testu myšího lymfomu a při dávkách 20 až 200 mg/kg (odhadovaný ekvivalent u člověka 1,67 až 16,7 mg/kg, na základě úpravy tělesného povrchu pro dospělého 60 kg; 0,1 až 1 násobek maxima doporučená lidská 24hodinová dávka ribavirinu) v mikronukleárním testu na myších. Dominantní letální test u potkanů byl negativní, což ukazuje, že pokud se mutace vyskytly u potkanů, nebyly přenášeny prostřednictvím samčích gamet.
Zhoršení plodnosti
Ribavirin prokázal významné embryocidní a teratogenní účinky v dávkách výrazně nižších, než je doporučená dávka pro člověka, u všech druhů zvířat, u kterých byly provedeny adekvátní studie. Byly zaznamenány malformace lebky, patra, oka, čelisti, končetin, kostry a gastrointestinálního traktu. Výskyt a závažnost teratogenních účinků se zvyšovala s eskalací dávky léku. Přežívání plodů a potomků bylo sníženo. V konvenčních studiích embryotoxicity/teratogenity u potkanů a králíků byly pozorované hladiny dávek, při kterých nedochází k žádným účinkům, výrazně pod úrovní navrhovaného klinického použití (0,3 mg/kg/den pro potkana i králíka; přibližně 0,06násobek doporučené 24hodinové dávky pro člověka ribavirin). Ve studii peri/postnatální toxicity u potkanů, kterým byla perorálně podávána dávka až 1 mg/kg/den (odhadovaná ekvivalentní dávka u člověka 0,17 mg/kg na základě úpravy tělesného povrchu pro dospělého o hmotnosti 60 kg, nebyla pozorována žádná mateřská toxicita ani účinky na potomstvo) přibližně 0,01násobek maximální doporučené lidské 24hodinové dávky ribavirinu) [viz KONTRAINDIKACE , a VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Fertilní ženy a partneři fertilních žen by neměli dostávat REBETOL, pokud pacientka a její partner nepoužívají účinnou antikoncepci (dvě spolehlivé formy). Na základě vícedávkového poločasu (t1/2) ribavirinu 12 dnů je nutné používat účinnou antikoncepci po dobu 6 měsíců po léčbě (např. 15 poločasů clearance ribavirinu).
REBETOL by měl být používán s opatrností u fertilních mužů. Ve studiích na myších k vyhodnocení časového průběhu a reverzibilita ribavirinem indukované testikulární degenerace při dávkách 15 až 150 mg/kg/den (odhadovaný ekvivalent u člověka 1,25 až 12,5 mg/kg/den, na základě úpravy plochy povrchu těla pro 60 kg dospělý; 0,1-0,8násobek maximální lidské 24hodinové dávky ribavirinu) podávané po dobu 3 nebo 6 měsíců, objevily se abnormality ve spermatu. Po ukončení léčby bylo v podstatě úplné zotavení z testikulární toxicity vyvolané ribavirinem patrné během 1 nebo 2 cyklů spermatogeneze.
Použití u konkrétních populací
Těhotenství
Kategorie těhotenství X
[Vidět KONTRAINDIKACE , VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ , a Neklinická toxikologie ].
Ošetření a doléčení:
Potenciální riziko pro plod
Je známo, že ribavirin se hromadí v intracelulárních složkách, odkud je velmi pomalu odstraňován. Není známo, zda ribavirin obsažený ve spermatu bude mít potenciální teratogenní účinek na oplodnění vajíček. Ve studii na potkanech se dospělo k závěru, že dominantní letalita nebyla indukována ribavirinem v dávkách až 200 mg/kg po dobu 5 dnů (odhadované ekvivalentní dávky u člověka 7,14 až 28,6 mg/kg, na základě úpravy plochy povrchu těla pro 60 kg dospělých; až 1,7násobek maximální doporučené dávky ribavirinu pro člověka). Vzhledem k potenciálním lidským teratogenním účinkům ribavirinu by však pacienti mužského pohlaví měli být poučeni, aby učinili veškerá opatření, aby se vyhnuli riziku otěhotnění jejich partnerek.
Ženy ve fertilním věku by neměly dostávat REBETOL, pokud během léčby nepoužívají účinnou antikoncepci (dvě spolehlivé formy). Kromě toho by měla být po dobu 6 měsíců po léčbě používána účinná antikoncepce na základě poločasu více dávek (t1/2) ribavirinu 12 dní.
Pacienti mužského pohlaví a jejich partnerky musí během léčby přípravkem REBETOL a po dobu 6 měsíců po léčbě používat účinnou antikoncepci (dvě spolehlivé formy) (např. 15 poločasů vylučování ribavirinu z těla).
Ribavirin Pregnancy Registry byl zřízen ke sledování mateřsko-fetálních výsledků těhotenství u pacientek a partnerek mužských pacientů vystavených ribavirinu během léčby a po dobu 6 měsíců po ukončení léčby. Lékařům a pacientům se doporučuje, aby takové případy hlásili na telefonním čísle 1-800-593-2214.
Kojící matky
Není známo, zda se přípravek REBETOL vylučuje do lidského mléka. Vzhledem k potenciálu závažných nežádoucích účinků léku u kojených dětí by se mělo rozhodnout, zda přerušit kojení nebo odložit či přerušit léčbu přípravkem REBETOL.
Pediatrické použití
Bezpečnost a účinnost přípravku REBETOL 200 mg v kombinaci s PegIntronem nebyla stanovena u pediatrických pacientů mladších 3 let. Při léčbě přípravkem REBETOL/INTRON A je třeba při rozhodování o léčbě dětského pacienta vzít v úvahu známky progrese onemocnění, jako je zánět jater a fibróza, a také prognostické faktory odpovědi, genotyp HCV a virovou zátěž. Přínosy léčby by měly být porovnány s pozorovanými bezpečnostními zjištěními.
Údaje z dlouhodobého sledování u pediatrických pacientů naznačují, že REBETOL v kombinaci s PegIntronem nebo s INTRONem A může vyvolat inhibici růstu, která má za následek snížení výšky u některých pacientů (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ a NEŽÁDOUCÍ REAKCE ].
Sebevražedné myšlenky nebo pokusy se vyskytovaly častěji u pediatrických pacientů, především dospívajících, ve srovnání s dospělými pacienty (2,4 % vs. 1 %) během léčby a sledování mimo terapii [vidět VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]. Stejně jako u dospělých pacientů se u dětských pacientů vyskytly jiné psychiatrické nežádoucí reakce (např. deprese, emoční labilita, somnolence), anémie a neutropenie (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Geriatrické použití
Klinické studie léčby REBETOL/INTRON A nebo PegIntron nezahrnovaly dostatečný počet subjektů ve věku 65 let a více, aby bylo možné určit, zda reagují odlišně od mladších subjektů.
Je známo, že REBETOL 200 mg je v podstatě vylučován ledvinami a riziko toxických reakcí na tento lék může být vyšší u pacientů s poruchou funkce ledvin. Vzhledem k tomu, že starší pacienti mají často sníženou funkci ledvin, je třeba dávat pozor na výběr dávky. Je třeba sledovat renální funkce a podle toho upravit dávkování. REBETOL 200 mg by se neměl používat u pacientů s clearance kreatininu nižší než 50 ml/min (viz KONTRAINDIKACE ].
Obecně platí, že REBETOL 200 mg tobolky by měly být podávány starším pacientům opatrně, počínaje spodní hranicí dávkovacího rozmezí, což odráží vyšší frekvenci snížené jaterní a srdeční funkce a souběžného onemocnění nebo jiné lékové terapie. V klinických studiích měli starší jedinci vyšší frekvenci anémie (67 %) než mladší pacienti (28 %) (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Příjemci orgánových transplantací
Bezpečnost a účinnost INTRONu A a PegIntronu samostatně nebo v kombinaci s REBETOLem pro léčbu hepatitidy C u příjemců transplantovaných jater nebo jiných orgánů nebyla stanovena. V malém (n=16) jednocentrovém nekontrolovaném případu bylo selhání ledvin u příjemců renálního aloštěpu, kteří dostávali kombinovanou léčbu interferonem alfa a ribavirinem, častější, než se očekávalo z předchozích zkušeností centra s příjemci renálního aloštěpu, kteří nedostávali kombinovanou léčbu. Vztah renálního selhání k rejekci renálního aloštěpu není jasný.
Souběžná infekce HIV nebo HBV
Bezpečnost a účinnost přípravků PegIntron/REBETOL 200 mg a INTRON A/REBETOL pro léčbu pacientů s HCV koinfikovaných HIV nebo HBV nebyla stanovena.
PŘEDÁVKOVAT
předávkováním jsou omezené zkušenosti. Bylo hlášeno akutní požití až 20 g kapslí REBETOL, požití až 120 milionů jednotek INTRON A a subkutánní dávky přípravku INTRON A až 10krát vyšší než doporučené dávky. Primární účinky, které byly pozorovány, jsou zvýšený výskyt a závažnost nežádoucích účinků souvisejících s terapeutickým použitím INTRON A a REBETOL. Při podání jednotlivých subkutánních dávek přípravku INTRON A, které překračují doporučené dávkování, však byly hlášeny abnormality jaterních enzymů, selhání ledvin, krvácení a infarkt myokardu.
Neexistuje žádné specifické antidotum pro předávkování přípravkem INTRON A nebo REBETOL 200 mg a hemodialýza a peritoneální dialýza nejsou pro léčbu předávkování těmito látkami účinné.
KONTRAINDIKACE
Kombinovaná terapie REBETOL je kontraindikována u:
- ženy, které jsou těhotné. REBETOL 200 mg může způsobit poškození plodu při podání těhotné ženě. REBETOL je kontraindikován u žen, které jsou nebo mohou otěhotnět. Pokud se REBETOL používá během těhotenství nebo pokud pacientka otěhotní během užívání přípravku REBETOL 200 mg, pacientka by měla být informována o potenciálním riziku pro její plod [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ , Použití ve specifických populacích , a INFORMACE PRO PACIENTA ]
- muži, jejichž partnerky jsou těhotné
- pacienti se známými reakcemi přecitlivělosti, jako je Stevens-Johnsonův syndrom, toxická, epidermální nekrolýza a multiformní erytém na ribavirin nebo kteroukoli složku přípravku
- pacientů s autoimunitní hepatitidou
- pacienti s hemoglobinopatií (např. talasémie major, srpkovitá anémie)
- pacientů s clearance kreatininu nižší než 50 ml/min. [vidět Použití ve specifických populacích a KLINICKÁ FARMAKOLOGIE ]
- Současné podávání REBETOLu a didanosinu je kontraindikováno, protože je zvýšená expozice aktivnímu metabolitu didanosinu (dideoxyadenosin 5'-trifosfát). U pacientů užívajících didanosin v kombinaci s ribavirinem bylo hlášeno fatální selhání jater, stejně jako periferní neuropatie, pankreatitida a symptomatická hyperlaktatémie/laktátová acidóza (viz DROGOVÉ INTERAKCE ].
KLINICKÁ FARMAKOLOGIE
Mechanismus působení
Ribavirin je antivirotikum [viz Mikrobiologie ].
Farmakokinetika
Farmakokinetické vlastnosti po jednorázové a opakované dávce u dospělých jsou shrnuty v tabulce 11. Ribavirin byl po perorálním podání rychle a extenzivně absorbován. V důsledku first-pass metabolismu však byla absolutní biologická dostupnost v průměru 64 % (44 %). Po jednotlivých dávkách 200 až 1200 mg ribavirinu byl lineární vztah mezi dávkou a AUCtf (AUC od času nula do poslední měřitelné koncentrace). Vztah mezi dávkou a Cmax byl křivočarý a měl tendenci k asymptotě nad jednotlivými dávkami 400 až 600 mg.
Po opakovaném perorálním podání na základě AUC12h byla pozorována 6násobná akumulace ribavirinu v plazmě. Po perorálním podávání 600 mg dvakrát denně bylo ustáleného stavu dosaženo přibližně za 4 týdny, s průměrnými plazmatickými koncentracemi v ustáleném stavu 2200 ng/ml (37 %). Po přerušení podávání byl průměrný poločas 298 (30 %) hodin, což pravděpodobně odráží pomalou eliminaci z neplazmatických kompartmentů.
Účinek antacida na absorpci ribavirinu
Současné podávání tobolek REBETOL s antacidem obsahujícím hořčík, hliník a simethikon vedlo k 14% snížení průměrné AUCtf ribavirinu. Klinický význam výsledků této studie s jednorázovou dávkou není znám.
Distribuce tkání
Transport ribavirinu do neplazmatických kompartmentů byl nejrozsáhleji studován v červených krvinkách a bylo identifikováno, že primárně probíhá prostřednictvím ekvilibračního nukleosidového transportéru typu es. Tento typ přenašeče je přítomen prakticky na všech typech buněk a může představovat rozsáhlý distribuční objem. Ribavirin se neváže na plazmatické proteiny.
Metabolismus a vylučování
Ribavirin má dvě cesty metabolismu: (i) cestu reverzibilní fosforylace v buňkách s jádry; a (ii) degradační dráhu zahrnující deribosylaci a hydrolýzu amidu za vzniku metabolitu triazolkarboxylové kyseliny. Ribavirin a jeho metabolity triazolkarboxamid a kyselina triazolkarboxylová jsou vylučovány ledvinami. Po perorálním podání 600 mg 14C-ribavirinu bylo přibližně 61 % a 12 % radioaktivity eliminováno močí a stolicí za 336 hodin. Nezměněný ribavirin představoval 17 % podané dávky.
Zvláštní populace
Renální dysfunkce
Farmakokinetika ribavirinu byla hodnocena po podání jedné perorální dávky (400 mg) ribavirinu subjektům neinfikovaným HCV s různým stupněm renální dysfunkce. Průměrná hodnota AUCtf byla třikrát vyšší u subjektů s hodnotami clearance kreatininu mezi 10 až 30 ml/min ve srovnání s kontrolními subjekty (clearance kreatininu vyšší než 90 ml/min). U subjektů s hodnotami clearance kreatininu mezi 30 až 60 ml/min byla AUCtf dvakrát vyšší ve srovnání s kontrolními subjekty. Zdá se, že zvýšené AUCtf je způsobeno snížením renální a nerenální clearance u těchto subjektů. Fáze 3 studií účinnosti zahrnovala subjekty s hodnotami clearance kreatininu vyššími než 50 ml/min. Farmakokinetiku opakovaných dávek ribavirinu nelze u pacientů s renální dysfunkcí přesně předpovědět. Ribavirin není účinně odstraněn hemodialýzou.
Pacienti s clearance kreatininu nižší než 50 ml/min by neměli být léčeni REBETOLem (viz KONTRAINDIKACE ].
Jaterní dysfunkce
Účinek jaterní dysfunkce byl hodnocen po jednorázové perorální dávce ribavirinu (600 mg). Průměrné hodnoty AUCtf se významně nelišily u subjektů s mírnou, středně těžkou nebo těžkou jaterní dysfunkcí (Child-Pugh klasifikace A, B nebo C) ve srovnání s kontrolními subjekty. Průměrné hodnoty Cmax se však zvyšovaly se závažností jaterní dysfunkce a byly dvakrát vyšší u subjektů s těžkou jaterní dysfunkcí ve srovnání s kontrolními subjekty.
Starší pacienti
Farmakokinetická hodnocení u starších subjektů nebyla provedena.
Rod
Ve studii s jednorázovou dávkou u 18 mužů a 18 žen nebyly zaznamenány žádné klinicky významné farmakokinetické rozdíly.
Pediatričtí pacienti
Farmakokinetické vlastnosti více dávek pro REBETOL 200 mg tobolky a INTRON A u pediatrických pacientů s chronickou hepatitidou C ve věku od 5 do 16 let jsou shrnuty v tabulce 12. Farmakokinetika REBETOL 200 mg a INTRON A (normalizovaná dávka) je podobná u dospělých a dětské předměty.
Úplné farmakokinetické vlastnosti perorálního roztoku REBETOL nebyly u pediatrických pacientů stanoveny. Hodnoty Cmin ribavirinu byly podobné po podání REBETOL perorálního roztoku nebo REBETOL 200 mg tobolek během 48 týdnů léčby u pediatrických pacientů (ve věku 3 až 16 let).
Byla provedena klinická studie u pediatrických pacientů s chronickou hepatitidou C ve věku od 3 do 17 let, ve které byla hodnocena farmakokinetika přípravků PegIntron a REBETOL (tobolky a perorální roztok). U pediatrických pacientů, kteří dostávali dávku PegIntronu upravenou podle tělesného povrchu v dávce 60 mcg/m²/týden, se předpokládalo, že logaritmicky transformovaný poměr expozice během dávkovacího intervalu bude o 58 % [90% CI: 141 %, 177 %] vyšší než pozorováno u dospělých, kteří dostávali 1,5 mcg/kg/týden. Farmakokinetika přípravku REBETOL (normalizovaná dávka) v této studii byla podobná farmakokinetice uváděné v předchozí studii přípravku REBETOL 200 mg v kombinaci s přípravkem INTRON A u pediatrických pacientů au dospělých.
Vliv potravy na absorpci ribavirinu
AUCtf i Cmax se zvýšily o 70 %, když byly tobolky REBETOL podávány s jídlem s vysokým obsahem tuku (841 kcal, 53,8 g tuku, 31,6 g bílkovin a 57,4 g sacharidů) ve farmakokinetické studii s jednorázovou dávkou [viz DÁVKOVÁNÍ A PODÁVÁNÍ ].
Mikrobiologie
Mechanismus působení
Mechanismus, kterým ribavirin přispívá k jeho antivirové účinnosti na klinice, není zcela objasněn. Ribavirin má přímou antivirovou aktivitu v tkáňové kultuře proti mnoha RNA virům. Ribavirin zvyšuje frekvenci mutací v genomech několika virů a ribavirintrifosfát inhibuje HCV polymerázu v biochemické reakci.
Antivirová aktivita v buněčné kultuře
Antivirová aktivita ribavirinu v replikonu HCV není dobře známa a nebyla definována kvůli buněčné toxicitě ribavirinu. Přímá antivirová aktivita byla pozorována v tkáňové kultuře jiných RNA virů. Anti-HCV aktivita interferonu byla prokázána v buňce obsahující samoreplikující se HCV-RNS (HCV replikonové buňky) nebo HCV infekci.
Odpor
Genotypy HCV vykazují širokou variabilitu ve své odpovědi na terapii pegylovaným rekombinantním lidským interferonem/ribavirinem. Genetické změny spojené s variabilní odpovědí nebyly identifikovány.
Křížový odpor
Nebyla hlášena žádná zkřížená rezistence mezi pegylovanými/nepegylovanými interferony a ribavirinem.
Toxikologie zvířat a farmakologie
Dlouhodobé studie na myších a potkanech [18 až 24 měsíců; dávky 20 až 75 a 10 až 40 mg/kg/den, v daném pořadí (odhadované ekvivalentní dávky u člověka 1,67 až 6,25 a 1,43 až 5,71 mg/kg/den, v daném pořadí, na základě úpravy tělesného povrchu pro dospělého o hmotnosti 60 kg; přibližně 0,1 až 0,4násobek maximální lidské 24hodinové dávky ribavirinu)] prokázaly vztah mezi chronickou expozicí ribavirinu a zvýšeným výskytem vaskulárních lézí (mikroskopických krvácení) u myší. U potkanů se degenerace sítnice objevila u kontrol, ale výskyt byl zvýšený u potkanů léčených ribavirinem.
Ve studii, ve které byl mláďatům potkanů postnatálně podáván ribavirin v dávkách 10, 25 a 50 mg/kg/den, došlo k úmrtí souvisejícím s lékem při dávce 50 mg/kg (při plazmatických koncentracích potkaních mláďat nižších, než jsou koncentrace v lidské plazmě u lidí terapeutická dávka) mezi 13. a 48. dnem studie. Mláďata potkanů, kterým byla podávána dávka od 7. do 63. postnatálního dne, prokázala menší, na dávce závislé snížení celkového růstu při všech dávkách, což se následně projevilo jako mírné snížení tělesné hmotnosti, délky temeno-zadní kosti, a délku kostí. Tyto účinky prokázaly reverzibilitu a nebyly pozorovány žádné histopatologické účinky na kost. Nebyly pozorovány žádné účinky ribavirinu na neurobehaviorální nebo reprodukční vývoj.
Klinické studie
Klinická studie 1 hodnotila monoterapii PegIntronem. Vidět Označení PegIntron pro informace o této zkoušce.
Kombinovaná terapie REBETOL/PegIntron
Dospělé subjekty
Studium 2
Randomizovaná studie porovnávala léčbu dvěma režimy PegIntron/REBETOL 200 mg [PegIntron 1,5 mcg/kg subkutánně jednou týdně/REBETOL 800 mg perorálně denně (v rozdělených dávkách); PegIntron 1,5 mcg/kg subkutánně jednou týdně po dobu 4 týdnů, poté 0,5 mcg/kg subkutánně jednou týdně po dobu 44 týdnů/REBETOL 1000 nebo 1200 mg perorálně denně (v rozdělených dávkách)] s INTRONem A [3 MIU subkutánně třikrát týdně/REBETOL10000 1200 mg perorálně denně (v rozdělených dávkách)] u 1530 dospělých s chronickou hepatitidou C. Subjekty dosud neléčené interferonem byly léčeny po dobu 48 týdnů a sledovány po dobu 24 týdnů po léčbě. Vhodní jedinci měli kompenzované onemocnění jater, detekovatelnou HCV-RNA, zvýšenou ALT a jaterní histopatologii konzistentní s chronickou hepatitidou.
Odpověď na léčbu byla definována jako nedetekovatelná HCV-RNA 24 týdnů po léčbě (viz tabulka 13). Míra odpovědi na dávku PegIntron 1,5 mcg/kg a ribavirin 800 mg byla vyšší než míra odpovědi na INTRON A/REBETOL (viz tabulka 13). Míra odpovědi na PegIntron 1,5→0,5 mcg/kg/REBETOL 200 mg byla v podstatě stejná jako odpověď na INTRON A/REBETOL (data neuvedena).
Jedinci s virovým genotypem 1, bez ohledu na virovou zátěž, měli nižší míru odezvy na PegIntron (1,5 mcg/kg)/REBETOL (800 mg) ve srovnání se subjekty s jinými virovými genotypy. Pacienti s oběma špatnými prognostickými faktory (genotyp 1 a vysoká virová nálož) měli míru odpovědi 30 % (78/256) ve srovnání s mírou odpovědi 29 % (71/247) při kombinované léčbě INTRON A/REBETOL.
Subjekty s nižší tělesnou hmotností měly tendenci mít vyšší míru nežádoucích reakcí [viz NEŽÁDOUCÍ REAKCE a vyšší míra odpovědi než u jedinců s vyšší tělesnou hmotností. Rozdíly v míře odpovědi mezi léčebnými rameny se podstatně nelišily s tělesnou hmotností.
Míra léčebné odpovědi na kombinovanou terapii PegIntron/REBETOL byla 49 % u mužů a 56 % u žen. Míra odpovědí byla nižší u Afroameričanů a Hispánců a vyšší u Asiatů ve srovnání s bělochy. Ačkoli Afroameričané měli vyšší podíl špatných prognostických faktorů ve srovnání s bělochy, počet nebělošských studovaných (11 % z celkového počtu) byl nedostatečný k tomu, aby umožnil smysluplné závěry o rozdílech v míře odpovědi po úpravě o prognostické faktory v této studii.
Jaterní biopsie byly získány před a po léčbě u 68 % subjektů. Ve srovnání s výchozí hodnotou bylo pozorováno přibližně u dvou třetin subjektů ve všech léčebných skupinách mírné snížení zánětu.
Studium 3
Ve velké komunitní studii ve Spojených státech bylo 4 913 subjektů s chronickou hepatitidou C randomizováno k podávání PegIntronu 1,5 mcg/kg subkutánně jednou týdně v kombinaci s 200 mg REBETOL dávkou 800 až 1 400 mg (dávkování podle hmotnosti [WBD]) nebo 800 mg (plochá) perorálně denně (v rozdělených dávkách) po dobu 24 nebo 48 týdnů na základě genotypu. Odpověď na léčbu byla definována jako nedetekovatelná HCV-RNA (na základě testu s dolním limitem detekce 125 IU/ml) 24 týdnů po léčbě.
Léčba PegIntronem 1,5 mcg/kg a REBETOLem 800 až 1400 mg vedla k vyšší setrvalé virologické odpovědi ve srovnání s PegIntronem v kombinaci s rovnou 800 mg denní dávkou REBETOLu. Subjekty s hmotností vyšší než 105 kg dosáhly největšího přínosu WBD, ačkoli mírný přínos byl také pozorován u subjektů s hmotností vyšší než 85 až 105 kg (viz tabulka 14). Přínos WBD u subjektů vážících více než 85 kg byl pozorován u HCV genotypů 1-3. K vyvození závěrů ohledně jiných genotypů nebyly k dispozici dostatečné údaje. Použití WBD vedlo ke zvýšenému výskytu anémie [viz NEŽÁDOUCÍ REAKCE ].
Celkem 1552 subjektů s hmotností vyšší než 65 kg ve studii 3 mělo genotyp 2 nebo 3 a byli randomizováni k 24 nebo 48týdenní léčbě. Při delší době léčby nebyl pozorován žádný další přínos.
Studium 4
Velká randomizovaná studie porovnávala bezpečnost a účinnost léčby po dobu 48 týdnů se dvěma režimy PegIntron/REBETOL 200 mg [PegIntron 1,5 mcg/kg a 1 mcg/kg subkutánně jednou týdně oba v kombinaci s REBETOL 800 až 1400 mg PO denně (ve dvou rozdělených dávky)] a Pegasys 180 mcg subkutánně jednou týdně v kombinaci s Copegusem 1000 až 1200 mg PO denně (ve dvou rozdělených dávkách) u 3070 dosud neléčených dospělých s chronickou hepatitidou C genotypu 1. V této studii nedostatek časné virologické odpovědi (nezjistitelné HCV-RNA nebo větší nebo rovné 2 log10 snížení od výchozí hodnoty) léčbou 12. týden byl kritériem pro přerušení léčby. SVR byla definována jako nedetekovatelná HCV-RNA (test Roche COBAS TaqMan, spodní limit kvantifikace 27 IU/ml) 24 týdnů po léčbě (viz tabulka 15).
Celková míra SVR byla u všech tří léčebných skupin podobná. Bez ohledu na léčebnou skupinu byly četnosti SVR nižší u subjektů se špatnými prognostickými faktory. Jedinci se špatnými prognostickými faktory randomizovaní k PegIntronu (1,5 mcg/kg)/REBETOL nebo Pegasys/Copegus však dosáhli vyšších hodnot SVR ve srovnání s podobnými subjekty randomizovanými k PegIntronu 1 mcg/kg/REBETOL. Pro PegIntron 1,5 mcg/kg a dávku REBETOL byly míry SVR pro subjekty s a bez následujících prognostických faktorů následující: cirhóza (10 % vs. 42 %), normální hladiny ALT (32 % vs. 42 %), výchozí virové zátěž vyšší než 600 000 IU/ml (35 % vs. 61 %), věk 40 let a starší (38 % vs. 50 %) a afroamerická rasa (23 % vs. 44 %). U subjektů s nedetekovatelnou HCV-RNA ve 12. týdnu léčby, kteří dostávali PegIntron (1,5 mcg/kg)/REBETOL 200 mg, byla míra SVR 81 % (328/407).
Studie 5 – Kombinovaná terapie REBETOL/PegIntron u selhání předchozí léčby
nekomparativní studii bylo 2293 subjektů se středně těžkou až těžkou fibrózou, u kterých selhala předchozí léčba kombinací interferon alfa/ribavirin, znovu léčeno PegIntronem, 1,5 mcg/kg subkutánně, jednou týdně, v kombinaci s ribavirinem s upravenou hmotností. Mezi způsobilé subjekty patřili dříve nereagující (jedinci, kteří byli HCV-RNA pozitivní na konci minimálně 12 týdnů léčby) a předchozí relabující (subjekty, kteří byli HCVRNA negativní na konci minimálně 12 týdnů léčby a následně relabovali po ukončení léčby následovat). Subjekty, které byly negativní v týdnu 12, byly léčeny po dobu 48 týdnů a sledovány po dobu 24 týdnů po léčbě. Odpověď na léčbu byla definována jako nedetekovatelná HCV-RNA 24 týdnů po léčbě (měřeno pomocí výzkumného testu, limit detekce 125 IU/ml). Celková míra odpovědi byla 22 % (497/2293) (99% CI: 19,5, 23,9). Subjekty s následujícími charakteristikami měly menší pravděpodobnost, že budou mít prospěch z opětovné léčby: předchozí nereagování, předchozí léčba pegylovaným interferonem, významná přemosťující fibróza nebo cirhóza a infekce genotypu 1.
Míry setrvalé virologické odezvy po opakované léčbě podle základních charakteristik jsou shrnuty v tabulce 16.
Dosažení nedetekovatelné HCV-RNA ve 12. týdnu léčby bylo silným prediktorem SVR. V této studii 1470 (64 %) subjektů nedosáhlo nedetekovatelné HCV-RNA v léčebném týdnu 12 a bylo jim nabídnuto zařazení do dlouhodobých léčebných studií z důvodu nedostatečné odpovědi na léčbu. Z 823 (36 %) subjektů, u kterých byla HCV-RNA nedetekovatelná ve 12. týdnu léčby, měli ti infikovaní genotypem 1 SVR 48 % (245/507), s rozsahem odpovědí podle skóre fibrózy (F4-F2) 39-55 %. Jedinci infikovaní genotypem 2/3, kteří byli HCV-RNA nedetekovatelná v léčebném týdnu 12, měli celkovou SVR 70 % (196/281), s rozsahem odpovědí podle skóre fibrózy (F4-F2) 60-83 %. U všech genotypů bylo vyšší skóre fibrózy spojeno se sníženou pravděpodobností dosažení SVR.
Pediatrické předměty
Dříve neléčení dětští jedinci ve věku 3 až 17 let s kompenzovanou chronickou hepatitidou C a detekovatelnou HCV-RNA byli léčeni REBETOL 15 mg/kg denně a PegIntron 60 mcg/m² jednou týdně po dobu 24 nebo 48 týdnů na základě genotypu HCV a výchozí virové hodnoty zatížení. Všechny subjekty měly být sledovány po dobu 24 týdnů po léčbě. Celkem bylo léčeno 107 subjektů, z nichž 52 % byly ženy, 89 % bělochů a 67 % bylo infikováno HCV genotypem 1. Subjekty infikované genotypem 1, 4 nebo genotypem 3 s HCV-RNA větším nebo rovným 600 000 IU/ml dostávali 48 týdnů terapie, zatímco ti infikovaní genotypem 2 nebo genotypem 3 s HCV-RNA méně než 600 000 IU/ml dostávali 24 týdnů terapie. Výsledky pokusu jsou shrnuty v tabulce 17.
REBETOL/INTRON A Kombinovaná terapie
Dospělé subjekty
Dříve neléčené subjekty
Dospělí s kompenzovanou chronickou hepatitidou C a detekovatelnou HCV-RNA (hodnoceno centrální laboratoří pomocí výzkumného testu RT-PCR), kteří dříve nebyli léčeni alfa interferonem, byli zařazeni do dvou multicentrických, dvojitě zaslepených studií (americké a mezinárodní) a randomizováni tak, aby dostávali REBETOL 200 mg tobolky 1200 mg/den (1000 mg/den pro subjekty s hmotností nižší nebo rovnou 75 kg) a INTRON A 3 MIU třikrát týdně nebo INTRON A a placebo po dobu 24 nebo 48 týdnů, po nichž následovalo 24 týdnů sledování mimo terapii. Mezinárodní studie neobsahovala 24týdenní léčebnou větev INTRON A a placebo. Do studie v USA bylo zařazeno 912 subjektů, kteří na začátku byli 67 % muži, 89 % bělochy s průměrným Knodell HAI skóre (I+II+III) 7,5 a 72 % genotypu 1. Mezinárodní studie, prováděná v Evropě, Izrael , Kanada a Austrálie, zahrnulo 799 subjektů (65 % mužů, 95 % bělochů, průměrné Knodellovo skóre 6,8 a 58 % genotyp 1).
Výsledky pokusů jsou shrnuty v tabulce 18.
Ze subjektů, které nedosáhly HCV-RNA pod limitem detekce testu založeného na výzkumu do 24. týdne léčby REBETOL/INTRON A, méně než 5 % reagovalo na dalších 24 týdnů kombinované léčby.
Mezi subjekty s HCV genotypem 1 léčenými terapií REBETOL/INTRON A, kteří dosáhli HCV-RNA pod detekční limit testu založeného na výzkumu do 24 týdnů, měli ti, kteří byli randomizováni do 48týdenní léčby, vyšší virologické odpovědi ve srovnání s pacienty ve 24. týdenní léčebná skupina. U subjektů s HCV negenotypu 1 randomizovaných k léčbě REBETOL/INTRON A po dobu 48 týdnů ve srovnání s 24 týdny nebylo pozorováno žádné zvýšení míry odpovědi.
Předměty s relapsem
Subjekty s kompenzovanou chronickou hepatitidou C a detekovatelnou HCV-RNA (posouzené centrální laboratoří pomocí výzkumného testu RT-PCR), u kterých došlo k relapsu po jedné nebo dvou cyklech léčby interferonem (definované jako abnormální hladiny ALT v séru), byly zařazeny do dvou multicentrické, dvojitě zaslepené studie (americké a mezinárodní) a randomizované k užívání REBETOL 1 200 mg/den (1 000 mg/den pro subjekty s hmotností ≤ 75 kg) a INTRON A 3 MIU třikrát týdně nebo INTRON A a placebo po dobu 24 týdnů s následným 24 týdnů sledování mimo terapii. Do studie v USA bylo zařazeno 153 subjektů, které byly na začátku 67 % muži, 92 % bělochy s průměrným Knodell HAI skóre (I+II+III) 6,8 a 58 % genotypu 1. Mezinárodní studie, prováděná v Evropě, Izrael , Kanada a Austrálie, zahrnulo 192 subjektů (64 % mužů, 95 % bělochů, průměrné Knodellovo skóre 6,6 a 56 % genotyp 1). Výsledky pokusů jsou shrnuty v tabulce 19.
Virologické a histologické odpovědi byly podobné u mužů a žen v dříve neléčených studiích a studiích s relapsem.
Pediatrické předměty
Pediatričtí jedinci ve věku 3 až 16 let s kompenzovanou chronickou hepatitidou C a detekovatelnou HCV-RNA (hodnoceno centrální laboratoří pomocí výzkumného testu RT-PCR) byli léčeni přípravkem REBETOL 15 mg/kg denně a INTRON A 3 MIU/ m² třikrát týdně po dobu 48 týdnů s následným 24týdenním sledováním po ukončení léčby. Celkem bylo léčeno 118 subjektů, z nichž 57 % byli muži, 80 % bělochů a 78 % genotypu 1. Subjekty mladší 5 let dostávaly perorální roztok REBETOL a osoby ve věku 5 let nebo starší dostávaly buď perorální roztok REBETOL nebo kapsle.
Výsledky pokusů jsou shrnuty v tabulce 20.
Jedinci s virovým genotypem 1, bez ohledu na virovou nálož, měli nižší míru odpovědi na kombinovanou terapii INTRON A/REBETOL ve srovnání s jedinci s genotypem non-1, 36 % vs. 81 %. Jedinci s oběma špatnými prognostickými faktory (genotyp 1 a vysoká virová nálož) měli míru odpovědi 26 % (13/50).
INFORMACE PRO PACIENTA
Nebyly poskytnuty žádné informace. Podívejte se prosím na VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ sekce.