Arimidex 1mg Anastrozole Použití, vedlejší účinky a dávkování. Cena v internetové lékárně. Generické léky bez předpisu.
Co je Arimidex a jak se používá?
Arimidex 1 mg je lék na předpis používaný k léčbě příznaků rakoviny prsu u žen po menopauze. Arimidex lze užívat samostatně nebo s jinými léky.
Arimidex patří do třídy léků nazývaných Antineoplastika, Inhibitor aromatázy.
Není známo, zda je přípravek Arimidex bezpečný a účinný u dětí.
Jaké jsou možné vedlejší účinky přípravku Arimidex?
Arimidex může způsobit závažné nežádoucí účinky včetně:
- dušnost,
- zlomenina kosti,
- oteklé žlázy,
- nevolnost,
- bolest v horní části žaludku,
- svědění,
- únava,
- ztráta chuti k jídlu,
- tmavá moč,
- stolice hliněné barvy,
- zežloutnutí kůže nebo očí (žloutenka),
- náhlá necitlivost nebo slabost (zejména na jedné straně těla),
- náhlá silná bolest hlavy,
- nezřetelná řeč,
- problémy s viděním nebo rovnováhou,
- horečka,
- bolest krku,
- otok v obličeji nebo na jazyku,
- pálení ve tvých očích,
- bolest kůže a
- červená nebo fialová kožní vyrážka, která se šíří (zejména v obličeji nebo horní části těla) s puchýři a olupováním
Pokud máte některý z výše uvedených příznaků, okamžitě vyhledejte lékařskou pomoc.
Mezi nejčastější vedlejší účinky přípravku Arimidex patří:
- slabost,
- návaly horka,
- pocit necitlivosti nebo brnění na kůži,
- otoky kotníků nebo nohou,
- bolest nebo ztuhlost kloubů,
- problémy s prsty při úchopu,
- bolest krku,
- bolest hlavy,
- bolesti zad,
- bolest kostí,
- Deprese,
- změny nálady,
- problémy se spánkem (nespavost),
- vysoký krevní tlak,
- těžká bolest hlavy,
- rozmazané vidění,
- bušení do krku nebo uší,
- nevolnost,
- zvracení a
- mírná vyrážka
Informujte lékaře, pokud máte jakýkoli nežádoucí účinek, který vás obtěžuje nebo který neustupuje.
Toto nejsou všechny možné vedlejší účinky přípravku Arimidex. Pro více informací se zeptejte svého lékaře nebo lékárníka.
Zavolejte svého lékaře o radu ohledně nežádoucích účinků. Nežádoucí účinky můžete hlásit úřadu FDA na čísle 1-800-FDA-1088.
POPIS
Tablety ARIMIDEX (anastrozol) pro perorální podání obsahují 1 mg anastrozolu, nesteroidního inhibitoru aromatázy. Chemicky je popsán jako 1,3-benzendiacetonitril, a, a, a', a'-tetramethyl-5-(1H-1,2,4-triazol-1-ylmethyl). Jeho molekulový vzorec je C17H19N5 a jeho strukturní vzorec je:
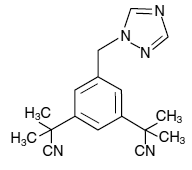
Anastrozol je téměř bílý prášek s molekulovou hmotností 293,4. Anastrozol má střední rozpustnost ve vodě (0,5 mg/ml při 25 °C); rozpustnost je nezávislá na pH ve fyziologickém rozmezí. Anastrozol je volně rozpustný v methanolu, acetonu, ethanolu a tetrahydrofuranu a velmi dobře rozpustný v acetonitrilu.
Každá tableta obsahuje jako neaktivní složky: laktózu, stearát hořečnatý, hydroxypropylmethylcelulózu, polyethylenglykol, povidon, sodnou sůl glykolátu škrobu a oxid titaničitý.
INDIKACE
Adjuvantní léčba
ARIMIDEX je indikován k adjuvantní léčbě postmenopauzálních žen s časným karcinomem prsu s pozitivními hormonálními receptory.
Léčba první linie
ARIMIDEX 1 mg je indikován k léčbě první linie u postmenopauzálních žen s lokálně pokročilým nebo metastatickým karcinomem prsu s pozitivním hormonálním receptorem nebo neznámým hormonálním receptorem.
Léčba druhé linie
ARIMIDEX 1 mg je indikován k léčbě pokročilého karcinomu prsu u postmenopauzálních žen s progresí onemocnění po léčbě tamoxifenem. Pacienti s ER-negativním onemocněním a pacienti, kteří nereagovali na předchozí léčbu tamoxifenem, reagovali na ARIMIDEX zřídka.
DÁVKOVÁNÍ A PODÁVÁNÍ
Doporučená dávka
Dávka přípravku ARIMIDEX je jedna 1 mg tableta užívaná jednou denně. U pacientek s pokročilým karcinomem prsu by ARIMIDEX 1 mg měl pokračovat až do progrese nádoru. ARIMIDEX lze užívat s jídlem nebo bez jídla.
Pro adjuvantní léčbu časného karcinomu prsu u postmenopauzálních žen není optimální délka terapie známa. Ve studii ATAC byl ARIMIDEX 1 mg podáván po dobu pěti let [viz Klinické studie ].
U pacientů s poruchou funkce ledvin nebo u starších pacientů není nutná žádná úprava dávkování (viz Použití u konkrétních populací ].
Pacienti s poruchou funkce jater
U pacientů s mírnou až středně těžkou poruchou funkce jater se nedoporučují žádné změny dávky. ARIMIDEX nebyl studován u pacientů s těžkou poruchou funkce jater (viz Použití ve specifických populacích ].
JAK DODÁVÁNO
Dávkové formy A Síly
Tablety jsou bílé, bikonvexní, potahované s obsahem 1 mg anastrozolu. Tablety jsou na jedné straně vyraženy logem skládajícím se z písmene „A“ (velké písmeno) se šipkou připojenou k úpatí prodloužené pravé nohy písmene „A“ a na zadní straně s označením síly tablety „Adx 1 “.
Skladování A Manipulace
Tyto tablety jsou dodávány v lahvičkách po 30 tabletách ( NDC 0310-0201-30).
Úložný prostor
Skladujte při kontrolované pokojové teplotě, 20-25 °C (68-77 °F) [viz USP ].
Distribuuje: AstraZeneca Pharmaceuticals LP Wilmington, DE 19850. Revize: květen 2014
VEDLEJŠÍ EFEKTY
Závažné nežádoucí účinky přípravku ARIMIDEX vyskytující se u méně než 1 z 10 000 pacientů jsou: 1) kožní reakce, jako jsou léze, vředy nebo puchýře; 2) alergické reakce s otokem obličeje, rtů, jazyka a/nebo hrdla. To může způsobit potíže s polykáním a/nebo dýcháním; a 3) změny v krevních testech jaterních funkcí, včetně zánětu jater s příznaky, které mohou zahrnovat celkový pocit špatného zdraví, se žloutenkou nebo bez ní, bolest jater nebo otok jater.
Časté nežádoucí účinky (vyskytující se s incidencí ≥ 10 %) u žen užívajících ARIMIDEX 1 mg zahrnovaly: návaly horka, astenie, artritidu, bolest, artralgii, hypertenzi, depresi, nevolnost a zvracení, vyrážku, osteoporózu, zlomeniny, bolest zad, nespavost, bolest hlavy, bolest kostí, periferní edém, zvýšený kašel, dušnost, faryngitida a lymfedém.
Ve studii ATAC byly nejčastěji hlášeným nežádoucím účinkem (> 0,1 %) vedoucím k přerušení léčby u obou léčebných skupin návaly horka, i když ve skupině ARIMIDEX 1 mg bylo méně pacientů, kteří přerušili léčbu v důsledku návalů horka.
Vzhledem k tomu, že klinické studie jsou prováděny za velmi odlišných podmínek, nelze míry nežádoucích reakcí pozorované v klinických studiích léku přímo srovnávat s mírami v klinických studiích jiného léku a nemusí odrážet míry pozorované v praxi.
Zkušenosti z klinických studií
Adjuvantní terapie
Údaje o nežádoucích účincích adjuvantní terapie jsou založeny na studii ATAC [viz Klinické studie ]. Medián trvání adjuvantní léčby pro hodnocení bezpečnosti byl 59,8 měsíce a 59,6 měsíce u pacientek užívajících ARIMIDEX 1 mg a tamoxifen 20 mg, v daném pořadí.
Nežádoucí účinky vyskytující se s výskytem alespoň 5 % v obou léčebných skupinách během léčby nebo do 14 dnů po ukončení léčby jsou uvedeny v tabulce 1.
Některé nežádoucí účinky a kombinace nežádoucích účinků byly prospektivně specifikovány pro analýzu na základě známých farmakologických vlastností a profilů vedlejších účinků těchto dvou léčiv (viz tabulka 2).
Ischemické kardiovaskulární příhody
Mezi léčebnými rameny v celkové populaci 6186 pacientů nebyl statistický rozdíl v ischemických kardiovaskulárních příhodách (4% ARIMIDEX vs. 3% tamoxifen).
V celkové populaci byla angina pectoris hlášena u 71/3092 (2,3 %) pacientů ve skupině ARIMIDEX 1 mg a 51/3094 (1,6 %) pacientů ve skupině s tamoxifenem; infarkt myokardu byl hlášen u 37/3092 (1,2 %) pacientů v rameni ARIMIDEX 1 mg a 34/3094 (1,1 %) pacientů v rameni s tamoxifenem.
U žen s preexistující ischemickou chorobou srdeční 465/6186 (7,5 %) byla incidence ischemických kardiovaskulárních příhod 17 % u pacientek užívajících ARIMIDEX a 10 % u pacientek užívajících tamoxifen. V této populaci pacientů byla angina pectoris hlášena u 25/216 (11,6 %) pacientů užívajících ARIMIDEX a 13/249 (5,2 %) pacientů užívajících tamoxifen; infarkt myokardu byl hlášen u 2/216 (0,9 %) pacientů užívajících ARIMIDEX a 8/249 (3,2 %) pacientů užívajících tamoxifen.
Zjištění hustoty kostních minerálů
Výsledky z kostní podstudie studie ATAC ve 12. a 24. měsíci prokázaly, že pacienti užívající ARIMIDEX 1 mg měli průměrný pokles jak v bederní páteři, tak v celkové minerální hustotě kyčelních kostí (BMD) ve srovnání s výchozí hodnotou. Pacientky užívající tamoxifen měly průměrné zvýšení BMD v oblasti bederní páteře a celkové kyčle ve srovnání s výchozí hodnotou.
Protože ARIMIDEX snižuje hladiny cirkulujícího estrogenu, může způsobit snížení minerální hustoty kostí.
Postmarketingová studie hodnotila kombinované účinky přípravku ARIMIDEX 1 mg a bisfosfonát risedronátu na změny BMD a markerů kostní resorpce a tvorby kostní tkáně od výchozích hodnot u postmenopauzálních žen s časným karcinomem prsu s pozitivními hormonálními receptory. Všichni pacienti dostávali suplementaci vápníkem a vitaminem D. Po 12 měsících bylo u pacientů, kteří neužívali bisfosfonáty, zaznamenáno malé snížení kostní denzity bederní páteře. Léčba bisfosfonáty zachovala kostní denzitu u většiny pacientů s rizikem zlomeniny.
postmenopauzálních žen s časným karcinomem prsu, u nichž je plánována léčba přípravkem ARIMIDEX 1 mg, by měl být stav kostí řízen podle pokynů pro léčbu, které jsou již k dispozici pro postmenopauzální ženy s podobným rizikem zlomenin z křehkosti.
Cholesterol
Během studie ATAC bylo u více pacientů užívajících ARIMIDEX 1 mg hlášeno zvýšení sérového cholesterolu ve srovnání s pacientkami užívajícími tamoxifen (9 % oproti 3,5 %, v tomto pořadí).
Postmarketingová studie také hodnotila jakékoli potenciální účinky přípravku ARIMIDEX na lipidový profil. V populaci primární analýzy pro lipidy (ARIMIDEX 1 mg samotný) nedošlo ke klinicky významné změně LDL-C od výchozí hodnoty do 12 měsíců a HDL-C od výchozí hodnoty do 12 měsíců.
V sekundární populaci pro lipidy (ARIMIDEX+risedronát) také nedošlo ke klinicky významné změně LDL-C a HDL-C od výchozí hodnoty do 12 měsíců.
obou populacích pro lipidy nebyl žádný klinicky významný rozdíl v celkovém cholesterolu (TC) nebo sérových triglyceridech (TG) po 12 měsících ve srovnání s výchozí hodnotou.
V této studii měla léčba po dobu 12 měsíců samotným ARIMIDEXEM 1 mg neutrální účinek na lipidový profil. Kombinovaná léčba ARIMIDEXEM a risedronátem měla také neutrální účinek na lipidový profil.
Studie poskytuje důkazy, že postmenopauzální ženy s časným karcinomem prsu, které mají být léčeny přípravkem ARIMIDEX 1 mg, by měly být léčeny podle aktuálních pokynů Národního vzdělávacího programu o cholesterolu pro řízení kardiovaskulárního rizika u jednotlivých pacientek se zvýšením LDL.
Jiné nežádoucí účinky
pacientů užívajících ARIMIDEX došlo ve srovnání s pacientkami užívajícími tamoxifen ke zvýšenému výskytu poruch kloubů (včetně artritidy, artrózy a artralgie). U pacientů užívajících ARIMIDEX došlo ke zvýšení incidence všech zlomenin (konkrétně zlomenin páteře, kyčle a zápěstí) [315 (10 %)] ve srovnání s pacientkami užívajícími tamoxifen [209 (7 %)].
Pacienti užívající ARIMIDEX 1 mg měli vyšší výskyt syndromu karpálního tunelu [78 (2,5 %)] ve srovnání s pacientkami užívajícími tamoxifen [22 (0,7 %)].
Vaginální krvácení se vyskytovalo častěji u pacientek léčených tamoxifenem oproti pacientkám léčeným ARIMIDEXEM 317 (10 %) oproti 167 (5 %).
U pacientek užívajících ARIMIDEX byl ve srovnání s pacientkami užívajícími tamoxifen nižší výskyt návalů horka, vaginálního krvácení, vaginálního výtoku, karcinomu endometria, žilních tromboembolických příhod a ischemických cerebrovaskulárních příhod.
10letý medián následných bezpečnostních výsledků ze studie ATAC
Výsledky jsou v souladu s předchozími analýzami.
Závažné nežádoucí účinky byly podobné u přípravku ARIMIDEX (50 %) a tamoxifenu (51 %).
- Kardiovaskulární příhody byly v souladu se známými bezpečnostními profily přípravku ARIMIDEX a tamoxifenu.
- Kumulativní výskyt všech prvních zlomenin (jak vážných, tak nezávažných, vyskytujících se buď během léčby nebo po léčbě) byl vyšší ve skupině ARIMIDEX (15 %) ve srovnání se skupinou s tamoxifenem (11 %). Tento zvýšený počet prvních zlomenin během léčby nepokračoval v období sledování po léčbě.
- Kumulativní incidence nových primárních karcinomů byla podobná ve skupině ARIMIDEX 1 mg (13,7 %) ve srovnání se skupinou tamoxifenu (13,9 %). V souladu s předchozími analýzami byl karcinom endometria vyšší ve skupině tamoxifenu (0,8 %) ve srovnání se skupinou ARIMIDEX (0,2 %).
- Celkový počet úmrtí (během léčby nebo mimo ni) byl mezi léčebnými skupinami podobný. Ve skupině léčené tamoxifenem bylo více úmrtí souvisejících s rakovinou prsu než ve skupině léčené přípravkem ARIMIDEX 1 mg.
Terapie první linie
Nežádoucí účinky vyskytující se s výskytem alespoň 5 % v obou léčebných skupinách studií 0030 a 0027 během léčby nebo do 2 týdnů po ukončení léčby jsou uvedeny v tabulce 3.
Méně časté nežádoucí účinky hlášené u pacientů užívajících ARIMIDEX 1 mg l mg ve studii 0030 nebo ve studii 0027 byly podobné těm, které byly hlášeny u terapie druhé linie.
Na základě výsledků z terapie druhé linie a stanoveného bezpečnostního profilu tamoxifenu byly statisticky analyzovány výskyty 9 předem specifikovaných kategorií nežádoucích účinků, které jsou potenciálně kauzálně spojené s jednou nebo oběma terapiemi kvůli jejich farmakologii. Mezi léčebnými skupinami nebyly pozorovány žádné významné rozdíly.
Terapie druhé linie
ARIMIDEX 1 mg byl tolerován ve dvou kontrolovaných klinických studiích (tj. ve studiích 0004 a 0005), kdy méně než 3,3 % pacientů léčených přípravkem ARIMIDEX a 4,0 % pacientů léčených megestrolacetátem ukončilo léčbu kvůli nežádoucí reakci.
Hlavní nežádoucí reakcí častější u přípravku ARIMIDEX než u megestrolacetátu byl průjem. Nežádoucí účinky hlášené u více než 5 % pacientů v kterékoli z léčebných skupin v těchto dvou kontrolovaných klinických studiích, bez ohledu na kauzalitu, jsou uvedeny níže:
Tělo jako celek: chřipkový syndrom; horečka; bolest krku; nevolnost; náhodné zranění; infekce
Kardiovaskulární: hypertenze; tromboflebitida
Jaterní: Gamma GT se zvýšil; SGOT se zvýšil; SGPT zvýšená Hematologická: Anémie; leukopenie
Metabolické a nutriční: zvýšená alkalická fosfatáza; ztráta váhy
Průměrné hladiny celkového cholesterolu v séru se u pacientů užívajících ARIMIDEX zvýšily o 0,5 mmol/l. Bylo prokázáno, že k těmto změnám přispívá zvýšení LDL cholesterolu.
Muskuloskeletální: myalgie; artralgie; patologická zlomenina
Nervový: Spavost; zmatek; nespavost; úzkost; nervozita
Respirační: sinusitida; bronchitida; rýma
Kůže a přílohy: Řídnutí vlasů (alopecie); pruritus
Urogenitální: Infekce močových cest; bolest prsou
Výskyt následujících skupin nežádoucích reakcí potenciálně kauzálně souvisejících s jednou nebo oběma terapiemi kvůli jejich farmakologii byl statisticky analyzován: přírůstek hmotnosti, edém, tromboembolické onemocnění, gastrointestinální poruchy, návaly horka a vaginální suchost. Těchto šest skupin a nežádoucí účinky zachycené ve skupinách byly prospektivně definovány. Výsledky jsou uvedeny v tabulce níže.
Postmarketingové zkušenosti
Tyto nežádoucí účinky jsou hlášeny dobrovolně z populace nejisté velikosti. Proto není vždy možné spolehlivě odhadnout jejich četnost nebo stanovit kauzální vztah k expozici drogám. Při použití přípravku Arimidex po schválení bylo hlášeno následující:
- Hepatobiliární příhody včetně zvýšení alkalické fosfatázy, alaninaminotransferázy, aspartátaminotransferázy, gama-GT a bilirubinu; hepatitida
- Vyrážka včetně případů mukokutánních poruch, jako je erythema multiforme a Stevens-Johnsonův syndrom
- Případy alergických reakcí včetně angioedému, kopřivky a anafylaxe [viz KONTRAINDIKACE ]
- Myalgie, spouštěcí prst a hyperkalcémie (s nebo bez zvýšení parathormonu)
DROGOVÉ INTERAKCE
tamoxifen
Současné podávání anastrozolu a tamoxifenu pacientkám s karcinomem prsu snížilo plazmatickou koncentraci anastrozolu o 27 %. Současné podávání anastrozolu a tamoxifenu však neovlivnilo farmakokinetiku tamoxifenu nebo N-desmethyltamoxifenu. Při střední době sledování 33 měsíců neprokázala kombinace ARIMIDEX a tamoxifenu žádný přínos z hlediska účinnosti ve srovnání s tamoxifenem u všech pacientek ani v subpopulaci pozitivní na hormonální receptory. Toto léčebné rameno bylo ze studie vyřazeno [viz Klinické studie ]. Na základě klinických a farmakokinetických výsledků ze studie ATAC by se tamoxifen neměl podávat s anastrozolem.
Estrogen
S přípravkem ARIMIDEX 1 mg by se neměly používat terapie obsahující estrogeny, protože mohou snižovat jeho farmakologický účinek.
Warfarin
Ve studii provedené na 16 mužských dobrovolnících anastrozol nezměnil expozici (měřeno pomocí C max a AUC) a antikoagulační aktivitu (měřenou protrombinovým časem, aktivovaným parciálním tromboplastinovým časem a trombinovým časem) R- a S- warfarin.
Cytochrom P450 Na základě výsledků in vitro a in vivo je nepravděpodobné, že by současné podávání přípravku ARIMIDEX 1 mg ovlivňovalo jiné léky v důsledku inhibice cytochromu P450 [viz KLINICKÁ FARMAKOLOGIE ].
VAROVÁNÍ
Zahrnuto jako součást OPATŘENÍ sekce.
OPATŘENÍ
Ischemické kardiovaskulární příhody
U žen s preexistující ischemickou chorobou srdeční byla u přípravku ARIMIDEX ve studii ATAC pozorována zvýšená incidence ischemických kardiovaskulárních příhod (17 % pacientek na ARIMIDEX 1 mg a 10 % pacientek na tamoxifenu). Zvažte riziko a přínosy léčby přípravkem ARIMIDEX u pacientů s již existující ischemickou chorobou srdeční (viz NEŽÁDOUCÍ REAKCE ]
Kostní efekty
Výsledky z kostní podstudie studie ATAC ve 12. a 24. měsíci prokázaly, že u pacientů užívajících ARIMIDEX došlo k průměrnému poklesu jak v bederní páteři, tak v celkové minerální hustotě kyčelních kostí (BMD) ve srovnání s výchozí hodnotou. Pacientky užívající tamoxifen měly průměrné zvýšení BMD v oblasti bederní páteře a celkové kyčle ve srovnání s výchozí hodnotou. Zvažte sledování minerální hustoty kostí u pacientů léčených ARIMIDEXEM [viz NEŽÁDOUCÍ REAKCE ].
Cholesterol
Během studie ATAC bylo hlášeno, že více pacientů užívajících ARIMIDEX 1 mg mělo zvýšený cholesterol v séru ve srovnání s pacienty užívajícími tamoxifen (9 % oproti 3,5 %) [viz NEŽÁDOUCÍ REAKCE ].
Informace pro pacienty
Vidět Označení pacienta schválené FDA (PATIENT INFORMACE).
Těhotenství
Pacientky je třeba upozornit, že ARIMIDEX 1 mg může způsobit poškození plodu. Je třeba je také upozornit, že ARIMIDEX 1 mg není určen k použití u premenopauzálních žen; pokud tedy otěhotní, měly by přestat užívat ARIMIDEX a okamžitě kontaktovat svého lékaře.
Alergické (hypersenzitivní) reakce
Pacienti by měli být informováni o možnosti závažných alergických reakcí s otokem obličeje, rtů, jazyka a/nebo hrdla (angioedém), který může způsobit potíže s polykáním a/nebo dýcháním, a okamžitě vyhledat lékařskou pomoc.
Ischemické kardiovaskulární příhody
Pacienti s preexistující ischemickou chorobou srdeční by měli být informováni, že při užívání přípravku ARIMIDEX 1 mg byl ve srovnání s tamoxifenem pozorován zvýšený výskyt kardiovaskulárních příhod. Pokud se u pacientů objeví nová nebo zhoršující se bolest na hrudi nebo dušnost, měli by okamžitě vyhledat lékařskou pomoc.
Kostní efekty
Pacientky by měly být informovány, že ARIMIDEX snižuje hladinu estrogenu. To může vést ke ztrátě obsahu minerálů v kostech, což může snížit pevnost kostí. Možným důsledkem sníženého obsahu minerálů v kostech je zvýšení rizika zlomenin.
Cholesterol
Pacienti by měli být informováni, že při léčbě přípravkem ARIMIDEX může být pozorována zvýšená hladina cholesterolu.
Lechtání, mravenčení nebo necitlivost
Pacienti by měli být informováni, že pokud zaznamenají lechtání, brnění nebo necitlivost, měli by to oznámit svému poskytovateli zdravotní péče.
tamoxifen
Pacientky je třeba upozornit, aby neužívaly ARIMIDEX s tamoxifenem.
Vynechané dávky
Informujte pacienty, že pokud vynechají dávku, užijte ji, jakmile si vzpomenou. Pokud je téměř čas na další dávku, vynechejte vynechanou dávku a vezměte si další pravidelnou dávku. Pacienti by neměli užívat dvě dávky současně.
Neklinická toxikologie
Karcinogeneze, mutageneze, zhoršení plodnosti
Konvenční studie karcinogeneze u potkanů v dávkách 1,0 až 25 mg/kg/den (asi 10 až 243násobek denní maximální doporučené dávky pro člověka na bázi mg/m²) podávaných perorální sondou po dobu až 2 let odhalila zvýšení výskyt hepatocelulárního adenomu a karcinomu a děložních stromálních polypů u žen a adenomu štítné žlázy u mužů při vysokých dávkách. U žen bylo pozorováno zvýšení v závislosti na dávce ve výskytu hyperplazie vaječníků a dělohy. Při dávce 25 mg/kg/den byly plazmatické hladiny AUC0-24 hodin u potkanů 110 až 125krát vyšší než hladiny vykazované u dobrovolníků po menopauze při doporučené dávce. Samostatná studie karcinogenity u myší při perorálních dávkách 5 až 50 mg/kg/den (asi 24 až 243násobek denní maximální doporučené dávky pro člověka na základě mg/m²) po dobu až 2 let vyvolala zvýšení výskytu benigních nádory ovariálních stromálních, epiteliálních a granulózních buněk ve všech dávkách. U samic myší bylo také pozorováno na dávce závislé zvýšení výskytu ovariální hyperplazie. Tyto ovariální změny jsou považovány za specifické účinky inhibice aromatázy na hlodavce a mají pro člověka sporný význam. Incidence lymfosarkomu byla zvýšena u mužů a žen při vysokých dávkách. Při dávce 50 mg/kg/den byly plazmatické hladiny AUC u myší 35 až 40krát vyšší než hladiny vykazované u dobrovolníků po menopauze při doporučené dávce.
testech in vitro (testy na bakteriích Ames a E. coli, test genových mutací CHO-K1) se ARIMIDEX 1 mg neprokázal jako mutagenní ani klastogenní ani in vitro (chromozomové aberace v lidských lymfocytech) ani in vivo (mikronukleární test na potkanech) .
Perorální podávání anastrozolu samicím potkanů (od 2 týdnů před pářením do 7. dne březosti) vedlo k významnému výskytu neplodnosti a snížení počtu životaschopných březostí při dávce 1 mg/kg/den (asi 10násobek doporučené dávky pro člověka na základě mg/m² a 9krát vyšší než AUC0-24 hod zjištěná u dobrovolníků po menopauze při doporučené dávce). Preimplantační ztráta vajíček nebo plodu byla zvýšena při dávkách rovných nebo vyšších než 0,02 mg/kg/den (přibližně jedna pětina doporučené dávky pro člověka na základě mg/m²). Obnova plodnosti byla pozorována po 5týdenním období bez podávání, které následovalo po 3 týdnech podávání. Není známo, zda tyto účinky pozorované u samic potkanů svědčí pro zhoršenou fertilitu u lidí.
Studie s opakovanými dávkami u potkanů, kterým byl po dobu 6 měsíců podáván anastrozol v dávkách rovných nebo vyšších než 1 mg/kg/den (což produkovalo plazmatický anastrozol Cssmax a AUC 0-24 h, které byly 19krát a 9krát vyšší než příslušné hodnoty zjištěné po menopauze dobrovolníků v doporučené dávce) vedlo k hypertrofii vaječníků a přítomnosti folikulárních cyst. Kromě toho byly v 6měsíčních studiích u psích samic pozorovány hyperplastické dělohy, kterým byly podávány dávky rovné nebo vyšší než 1 mg/kg/den (což produkovalo plazmatický anastrozol Cssmax a AUC0-24 h, které byly 22krát a 16krát vyšší než u příslušných hodnoty zjištěné u postmenopauzálních žen při doporučené dávce). Není známo, zda jsou tyto účinky na reprodukční orgány zvířat spojeny s poruchou plodnosti u premenopauzálních žen.
Použití u konkrétních populací
Těhotenství
Kategorie těhotenství X [vidět KONTRAINDIKACE ]
ARIMIDEX může způsobit poškození plodu, když je podáván těhotné ženě a nenabízí žádný klinický přínos pro premenopauzální ženy s rakovinou prsu. ARIMIDEX je kontraindikován u žen, které jsou nebo mohou otěhotnět. Ve studiích na zvířatech způsobil anastrozol selhání těhotenství, zvýšenou ztrátu těhotenství a známky opožděného vývoje plodu. Neexistují žádné studie použití přípravku ARIMIDEX 1 mg u těhotných žen. Pokud se ARIMIDEX 1 mg používá během těhotenství nebo pokud pacientka otěhotní během užívání tohoto léku, pacientka by měla být informována o potenciálním riziku pro plod a potenciálním riziku ztráty těhotenství.
reprodukčních studiích na zvířatech dostávaly březí potkaní potkani a králíci anastrozol během organogeneze v dávkách rovných nebo vyšších než 1 (potkani) a 1/3 (králíci) doporučené dávky pro člověka na základě mg/m². U obou druhů anastrozol procházel placentou a došlo ke zvýšené ztrátě březosti (zvýšené předimplantační a/nebo postimplantační ztráty, zvýšená resorpce a snížený počet živých plodů). U potkanů byly tyto účinky závislé na dávce a hmotnosti placenty byly významně zvýšeny. Fetotoxicita, včetně opožděného vývoje plodu (tj. neúplná osifikace a snížená tělesná hmotnost plodu), se objevila u potkanů při dávkách anastrozolu, které vedly k maximálním plazmatickým hladinám 19krát vyšším než sérové hladiny u lidí při terapeutické dávce (AUC 0-24 hodin 9krát vyšší) . U králíků způsobil anastrozol selhání březosti v dávkách rovných nebo vyšších než 16násobek doporučené dávky u člověka na základě mg/m² [viz Toxikologie zvířat a/nebo farmakologie ].
Kojící matky
Není známo, zda se anastrozol vylučuje do lidského mateřského mléka. Vzhledem k tomu, že mnoho léků se vylučuje do lidského mateřského mléka a kvůli tumorigenicitě prokázané u anastrozolu ve studiích na zvířatech nebo kvůli možnosti závažných nežádoucích reakcí u kojených dětí, je třeba rozhodnout, zda přerušit kojení nebo přerušit podávání léku, přičemž je třeba vzít v úvahu význam léku pro matku.
Pediatrické použití
Klinické studie u dětských pacientů zahrnovaly placebem kontrolovanou studii u pubertálních chlapců dospívajícího věku s gynekomastií a jednoramennou studii u dívek s McCune-Albright syndromem a progresivní předčasnou pubertou. Účinnost přípravku ARIMIDEX 1 mg v léčbě pubertální gynekomastie u dospívajících chlapců a v léčbě předčasné puberty u dívek s McCune-Albright syndromem nebyla prokázána.
Studie gynekomastie
Randomizovaná, dvojitě zaslepená, placebem kontrolovaná, multicentrická studie zahrnovala 80 chlapců s pubertální gynekomastií ve věku 11 až 18 let. Pacienti byli randomizováni do denního režimu buď ARIMIDEX 1 mg, nebo placeba. Po 6 měsících léčby nebyl žádný statisticky významný rozdíl v procentu pacientek, u kterých došlo ke snížení gynekomastie o ≥ 50 % (primární analýza účinnosti). Sekundární analýzy účinnosti (absolutní změna objemu prsou, procento pacientek, u kterých došlo k jakémukoli snížení vypočteného objemu gynekomastie, vymizení bolesti prsu) byly v souladu s primární analýzou účinnosti. Koncentrace estradiolu v séru v 6. měsíci léčby byly sníženy o 15,4 % ve skupině ARIMIDEX a 4,5 % ve skupině s placebem.
Nežádoucí účinky, které zkoušející vyhodnotili jako související s léčbou, se objevily u 16,3 % pacientů léčených přípravkem ARIMIDEX a 8,1 % pacientů léčených placebem, přičemž nejčastější bylo akné (7 % ARIMIDEX a 2,7 % placebo) a bolest hlavy (7 % ARIMIDEX 1 mg a 0 % placebo); všechny ostatní nežádoucí účinky vykazovaly malé rozdíly mezi léčebnými skupinami. Jeden pacient léčený přípravkem ARIMIDEX 1 mg přerušil studii z důvodu zvětšení varlat. Průměrná základní hodnota odečtená změna objemu varlat po 6 měsících léčby byla + 6,6 ± 7,9 cm³ u pacientů léčených ARIMIDEXEM a + 5,2 ± 8,0 cm³ ve skupině s placebem.
Studie McCune-Albrightova syndromu
Multicentrická, jednoramenná, otevřená studie byla provedena u 28 dívek s McCune-Albrightovým syndromem a progresivní předčasnou pubertou ve věku od 2 do
pěti pacientů (18 %) se vyskytly nežádoucí účinky, které byly považovány za možné související s přípravkem ARIMIDEX. Jednalo se o nevolnost, akné, bolest v končetině, zvýšenou alanintransaminázu a aspartáttransaminázu a alergickou dermatitidu.
Farmakokinetika u dětských pacientů
Po opakovaném podání 1 mg jednou denně u pediatrických pacientů byla průměrná doba k dosažení maximální koncentrace anastrozolu 1 hodina. Průměrné (rozmezí) dispoziční parametry anastrozolu u pediatrických pacientů byly popsány CL/F 1,54 l/h (0,77-4,53 l/h) a V/F 98,4 l (50,7-330,0 l). Terminální eliminační poločas byl 46,8 h, což bylo podobné jako u žen po menopauze léčených anastrozolem pro karcinom prsu. Na základě populační farmakokinetické analýzy byla farmakokinetika anastrozolu podobná u chlapců s pubertální gynekomastií a dívek s McCune-Albright syndromem.
Geriatrické použití
Ve studiích 0030 a 0027 bylo asi 50 % pacientů ve věku 65 let nebo starších. Pacienti ve věku ≥ 65 let měli mírně lepší odpověď nádoru a dobu do progrese nádoru než pacienti ve věku
Ve studii ATAC bylo 45 % pacientů ve věku 65 let nebo starších. Účinnost přípravku ARIMIDEX 1 mg ve srovnání s tamoxifenem u pacientů ve věku 65 let nebo starších (N=1413 pro ARIMIDEX a N=1410 pro tamoxifen, poměr rizika pro přežití bez onemocnění byl 0,93 [95% CI: 0,80, 1,08]) byl nižší než účinnost pozorovaná u pacientů mladších 65 let (N=1712 pro ARIMIDEX a N=1706 pro tamoxifen, poměr rizika pro přežití bez onemocnění byl 0,79 [95% CI: 0,67, 0,94]).
Farmakokinetika anastrozolu není ovlivněna věkem.
Renální poškození
Vzhledem k tomu, že pouze asi 10 % anastrozolu se vylučuje v nezměněné podobě močí, porucha funkce ledvin neovlivňuje celkovou tělesnou clearance. Úprava dávkování u pacientů s poruchou funkce ledvin není nutná (viz DÁVKOVÁNÍ A PODÁVÁNÍ a KLINICKÁ FARMAKOLOGIE ].
Poškození jater
Plazmatické koncentrace anastrozolu u subjektů s jaterní cirhózou byly v rozmezí koncentrací pozorovaných u normálních subjektů ve všech klinických studiích. Úprava dávkování proto také není nutná u pacientů se stabilní cirhózou jater. ARIMIDEX nebyl studován u pacientů s těžkou poruchou funkce jater (viz DÁVKOVÁNÍ A PODÁVÁNÍ a KLINICKÁ FARMAKOLOGIE ].
PŘEDÁVKOVAT
Klinické studie byly provedeny s ARIMIDEXEM, až 60 mg v jedné dávce podávané zdravým mužským dobrovolníkům a až 10 mg denně podávaným ženám po menopauze s pokročilým karcinomem prsu; tyto dávky byly tolerovány. Jednorázová dávka přípravku ARIMIDEX 1 mg, která vede k život ohrožujícím příznakům, nebyla stanovena. Na předávkování neexistuje žádné specifické antidotum a léčba musí být symptomatická. Při léčbě předávkování vezměte v úvahu, že mohlo být užito více látek. Zvracení může být vyvoláno, pokud je pacient ve střehu. Dialýza může být užitečná, protože ARIMIDEX není silně vázán na proteiny. Je indikována obecná podpůrná péče, včetně častého sledování vitálních funkcí a pečlivého sledování pacienta.
KONTRAINDIKACE
Těhotenství A Premenopauzální ženy
ARIMIDEX 1 mg může způsobit poškození plodu, je-li podáván těhotné ženě a nenabízí žádný klinický přínos u premenopauzálních žen s rakovinou prsu. ARIMIDEX 1 mg je kontraindikován u žen, které jsou nebo mohou otěhotnět. Neexistují žádné adekvátní a dobře kontrolované studie u těhotných žen užívajících ARIMIDEX. Pokud se ARIMIDEX 1 mg používá během těhotenství nebo pokud pacientka otěhotní během užívání tohoto léku, pacientka by měla být informována o potenciálním riziku pro plod nebo potenciálním riziku ztráty těhotenství (viz Použití ve specifických populacích ].
Přecitlivělost
ARIMIDEX 1 mg je kontraindikován u všech pacientů, u kterých se projevila hypersenzitivní reakce na léčivo nebo na kteroukoli pomocnou látku. Pozorované reakce zahrnují anafylaxi, angioedém a kopřivku [viz NEŽÁDOUCÍ REAKCE ]
KLINICKÁ FARMAKOLOGIE
Mechanismus působení
Růst mnoha rakovin prsu je stimulován nebo udržován estrogeny.
postmenopauzálních žen jsou estrogeny odvozeny především z působení enzymu aromatázy, který přeměňuje adrenální androgeny (především androstendion a testosteron) na estron a estradiol. Potlačení biosyntézy estrogenu v periferních tkáních a v rakovinné tkáni samotné lze tedy dosáhnout specifickou inhibicí enzymu aromatázy.
Anastrozol je selektivní nesteroidní inhibitor aromatázy. Významně snižuje sérové koncentrace estradiolu a nemá detekovatelný účinek na tvorbu adrenálních kortikosteroidů nebo aldosteronu.
Farmakodynamika
Účinek na estradiol
Průměrné sérové koncentrace estradiolu byly hodnoceny v několika studiích s denním dávkováním 0,5, 1, 3, 5 a 10 mg přípravku ARIMIDEX u postmenopauzálních žen s pokročilým karcinomem prsu. U všech dávek byla pozorována klinicky významná suprese sérového estradiolu. Dávky 1 mg a vyšší vedly k supresi průměrných sérových koncentrací estradiolu na spodní hranici detekce (3,7 pmol/l). Doporučená denní dávka, ARIMIDEX 1 mg, snížila estradiol přibližně o 70 % během 24 hodin a přibližně o 80 % po 14 dnech denního dávkování. Suprese sérového estradiolu byla udržována po dobu až 6 dnů po ukončení denního podávání přípravku ARIMIDEX 1 mg.
Účinek přípravku ARIMIDEX 1 mg u premenopauzálních žen s časným nebo pokročilým karcinomem prsu nebyl studován. Protože aromatizace adrenálních androgenů není významným zdrojem estradiolu u premenopauzálních žen, neočekává se, že by ARIMIDEX snižoval hladiny estradiolu u premenopauzálních žen.
Účinek na kortikosteroidy
několika studiích s denním dávkováním 3, 5 a 10 mg byla selektivita anastrozolu hodnocena zkoumáním účinků na syntézu kortikosteroidů. U všech dávek anastrozol neovlivnil sekreci kortizolu nebo aldosteronu na začátku léčby nebo jako odpověď na ACTH. Není nutná žádná substituční léčba glukokortikoidy nebo mineralokortikoidy anastrozolem.
Další endokrinní účinky
V několika studiích s denním dávkováním 5 a 10 mg byl měřen hormon stimulující štítnou žlázu (TSH); během podávání ARIMIDEXu nedošlo ke zvýšení TSH. ARIMIDEX nemá přímou progestogenní, androgenní nebo estrogenní aktivitu u zvířat, ale narušuje cirkulující hladiny progesteronu, androgenů a estrogenů.
Farmakokinetika
Vstřebávání
Inhibice aktivity aromatázy je primárně způsobena anastrozolem, mateřským lékem. Absorpce anastrozolu je rychlá a maximální plazmatické koncentrace se typicky dosahují během 2 hodin po podání nalačno. Studie s radioaktivně značeným léčivem prokázaly, že perorálně podávaný anastrozol se dobře vstřebává do systémového oběhu. Jídlo snižuje rychlost, ale ne celkový rozsah absorpce anastrozolu. Průměrná C max anastrozolu se snížila o 16 % a medián Tmax se zpozdil ze 2 na 5 hodin, když byl anastrozol podán 30 minut po jídle. Farmakokinetika anastrozolu je lineární v rozmezí dávek 1 až 20 mg a při opakovaném podávání se nemění. Farmakokinetika anastrozolu byla u pacientek a zdravých dobrovolníků podobná.
Rozdělení
Plazmatické hladiny v ustáleném stavu jsou přibližně 3 až 4krát vyšší než hladiny pozorované po jedné dávce přípravku ARIMIDEX. Plazmatické koncentrace se blíží rovnovážnému stavu přibližně po 7 dnech podávání jednou denně. Anastrozol se v terapeutickém rozmezí váže na plazmatické proteiny ze 40 %.
Metabolismus
Metabolismus anastrozolu probíhá N-dealkylací, hydroxylací a glukuronidací. V lidské plazmě a moči byly identifikovány tři metabolity anastrozolu (triazol, glukuronidový konjugát hydroxyanastrozolu a glukuronidový konjugát samotného anastrozolu). Hlavní cirkulující metabolit anastrozolu, triazol, postrádá farmakologickou aktivitu.
Anastrozol inhiboval reakce katalyzované cytochromem P450 1A2, 2C8/9 a 3A4 in vitro s hodnotami Ki, které byly přibližně 30krát vyšší než průměrné hodnoty Cmax v ustáleném stavu pozorované po denní dávce 1 mg. Anastrozol neměl žádný inhibiční účinek na reakce katalyzované cytochromem P450 2A6 nebo 2D6 in vitro. Podání jednorázové dávky 30 mg/kg nebo více dávek 10 mg/kg anastrozolu zdravým subjektům nemělo žádný vliv na clearance antipyrinu nebo na obnovu antipyrinových metabolitů močí.
Vylučování
85 procent radioaktivně značeného anastrozolu bylo nalezeno ve stolici a moči. Metabolismus jater odpovídá přibližně 85 % eliminace anastrozolu. Renální eliminace představuje přibližně 10 % celkové clearance. Průměrný eliminační poločas anastrozolu je 50 hodin.
Vliv pohlaví a věku
Farmakokinetika anastrozolu byla zkoumána u dobrovolnic po menopauze a pacientek s rakovinou prsu. V rozmezí 80 let nebyly pozorovány žádné účinky související s věkem.
Vliv rasy
Hladiny estradiolu a estronsulfátu v séru byly podobné u japonských a kavkazských postmenopauzálních žen, které dostávaly 1 mg anastrozolu denně po dobu 16 dnů. Průměrné minimální plazmatické koncentrace anastrozolu v ustáleném stavu u bělošských a japonských postmenopauzálních žen byly 25,7 a 30,4 ng/ml.
Vliv poškození ledvin
Farmakokinetika anastrozolu byla zkoumána u subjektů s poruchou funkce ledvin. Renální clearance anastrozolu klesala úměrně s clearance kreatininu a byla přibližně o 50 % nižší u dobrovolníků s těžkou poruchou funkce ledvin (clearance kreatininu DÁVKOVÁNÍ A PODÁVÁNÍ a Použití ve specifických populacích ].
Vliv poškození jater
Farmakokinetika anastrozolu byla zkoumána u subjektů s jaterní cirhózou související se zneužíváním alkoholu. Zdánlivá perorální clearance (CL/F) anastrozolu byla přibližně o 30 % nižší u subjektů se stabilní cirhózou jater než u kontrolních subjektů s normální funkcí jater. Tyto plazmatické koncentrace však byly stále v rozmezí hodnot pozorovaných u normálních subjektů. Vliv těžkého poškození jater nebyl studován. U stabilní jaterní cirhózy není nutná žádná úprava dávky (viz DÁVKOVÁNÍ A PODÁVÁNÍ a Použití u konkrétních populací ].
Toxikologie zvířat a/nebo farmakologie
Reprodukční toxikologie
Bylo zjištěno, že anastrozol prochází placentou po perorálním podání 0,1 mg/kg u potkanů a králíků (přibližně 1 a 1,9násobek doporučené dávky pro člověka, v přepočtu na mg/m²). Studie na potkanech a králících v dávkách rovných nebo vyšších než 0,1 a 0,02 mg/kg/den, v daném pořadí (asi 1 a 1/3 doporučené dávky pro člověka na základě mg/m²), podávaných během období organogeneze ukázala, že anastrozol zvyšuje ztrátu těhotenství (zvýšená pre- a/nebo postimplantační ztráta, zvýšená resorpce a snížený počet živých plodů); účinky byly u potkanů závislé na dávce. Hmotnost placenty byla významně zvýšena u potkanů při dávkách 0,1 mg/kg/den nebo vyšších.
Důkazy fetotoxicity, včetně opožděného vývoje plodu (tj. neúplná osifikace a snížená tělesná hmotnost plodů), byly pozorovány u potkanů, kterým byly podávány dávky 1 mg/kg/den (což produkovalo plazmatický anastrozol Cssmax a AUC 0-24 hod., které byly 19násobné a 9krát vyšší než příslušné hodnoty zjištěné u dobrovolníků po menopauze při doporučené dávce). U potkanů, kterým byly podávány dávky až do 1,0 mg/kg/den, nebyla prokázána teratogenita. U králíků způsobil anastrozol selhání březosti v dávkách rovných nebo vyšších než 1,0 mg/kg/den (asi 16násobek doporučené dávky pro člověka na základě mg/m²); nebyla prokázána teratogenita u králíků, kterým byla podávána dávka 0,2 mg/kg/den (asi trojnásobek doporučené dávky pro člověka na základě mg/m²).
Klinické studie
Adjuvantní léčba rakoviny prsu u žen po menopauze
Multicentrická dvojitě zaslepená studie (ATAC) randomizovala 9 366 postmenopauzálních žen s operabilním karcinomem prsu k adjuvantní léčbě přípravkem ARIMIDEX 1 mg denně, tamoxifenem 20 mg denně nebo kombinací těchto dvou léčebných postupů po dobu pěti let nebo do recidivy onemocnění.
Primárním cílovým parametrem studie bylo přežití bez onemocnění (tj. doba do výskytu vzdálené nebo lokální recidivy nebo kontralaterálního karcinomu prsu nebo úmrtí z jakékoli příčiny). Sekundární cílové parametry studie zahrnovaly vzdálené přežití bez onemocnění, výskyt kontralaterálního karcinomu prsu a celkové přežití. Při střední době sledování 33 měsíců neprokázala kombinace ARIMIDEX a tamoxifenu žádný přínos z hlediska účinnosti ve srovnání s tamoxifenem u všech pacientek ani u subpopulace pozitivní na hormonální receptory. Tato léčebná větev byla ze studie vyřazena. Na základě klinických a farmakokinetických výsledků ze studie ATAC by se tamoxifen neměl podávat s anastrozolem [viz DROGOVÉ INTERAKCE ].
Demografické a další základní charakteristiky byly u všech tří léčebných skupin podobné (viz tabulka 7).
Pacienti ve dvou větvích monoterapie studie ATAC byli léčeni po dobu mediánu 60 měsíců (5 let) a sledováni po dobu mediánu 68 měsíců. Přežití bez onemocnění v populaci intent-to-treat bylo statisticky významně zlepšeno [Hazard Ratio (HR) = 0,87, 95% CI: 0,78, 0,97, p=0,0127] v rameni ARIMIDEX ve srovnání s ramenem s tamoxifenem. V subpopulaci s pozitivními hormonálními receptory, která představuje asi 84 % pacientů ve studii, se přežití bez onemocnění také statisticky významně zlepšilo (HR = 0,83, 95% CI: 0,73, 0,94, p = 0,0049) ve skupině ARIMIDEX 1 mg ve srovnání s ramenem tamoxifen rameno.
Obrázek 1: Kaplan Meierova křivka přežití bez onemocnění pro všechny pacienty randomizované k monoterapii ARIMIDEXEM 1 mg nebo tamoxifenem ve studii ATAC (Intent-to-Treat) 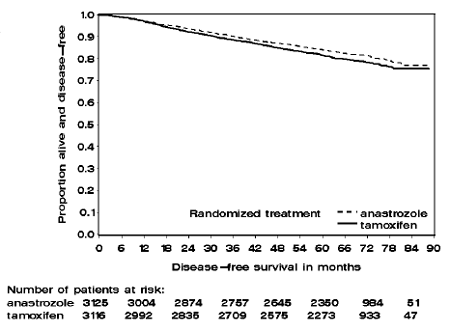
Obrázek 2: Přežití bez onemocnění u subpopulace pacientů s pozitivními hormonálními receptory randomizovaných k monoterapii ARIMIDEX nebo tamoxifenem ve studii ATAC 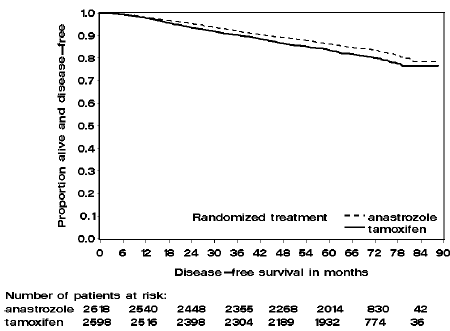
Údaje o přežití s 68měsíčním sledováním jsou uvedeny v tabulce 9.
Ve skupině pacientek, které měly předchozí adjuvantní chemoterapii (N=698 pro ARIMIDEX 1 mg a N=647 pro tamoxifen), byl poměr rizika pro přežití bez onemocnění 0,91 (95% CI: 0,73 až 1,13) ve skupině ARIMIDEX ve srovnání s tamoxifenové rameno.
Četnost jednotlivých příhod v populaci intent-to-treat a subpopulaci pozitivní na hormonální receptory je popsána v tabulce 8.
Shrnutí výsledků studie účinnosti je uvedeno v tabulce 9.
10letý medián následného sledování Výsledky účinnosti ze studie ATAC
V následné analýze studie ATAC byli pacienti ve dvou ramenech s monoterapií sledováni po dobu mediánu 120 měsíců (10 let). Pacienti dostávali studijní léčbu po dobu mediánu 60 měsíců (5 let) (viz tabulka 10).
Obrázek 3: Kaplan Meierova křivka přežití bez onemocnění pro všechny pacienty randomizované k monoterapii ARIMIDEX nebo tamoxifenem ve studii ATAC (intent-to-treat)a 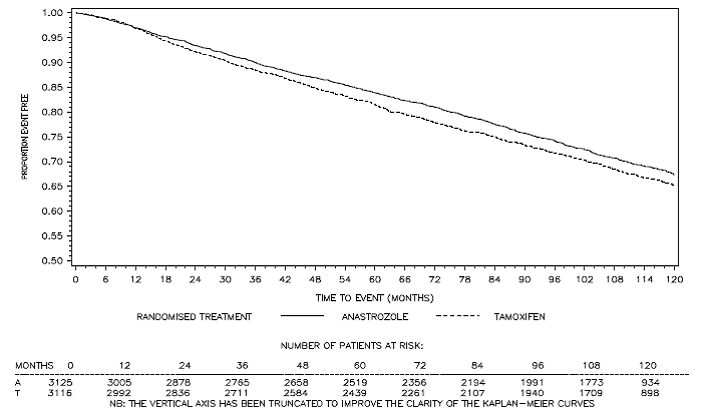
a Podíl pacientů se 120měsíčním sledováním byl 29,4 %.
Obrázek 4: Přežití bez onemocnění u subpopulace pacientů pozitivních na hormonální receptory randomizovaných k monoterapii ARIMIDEXEM 1 mg nebo tamoxifenem ve studii ATAC Trialb 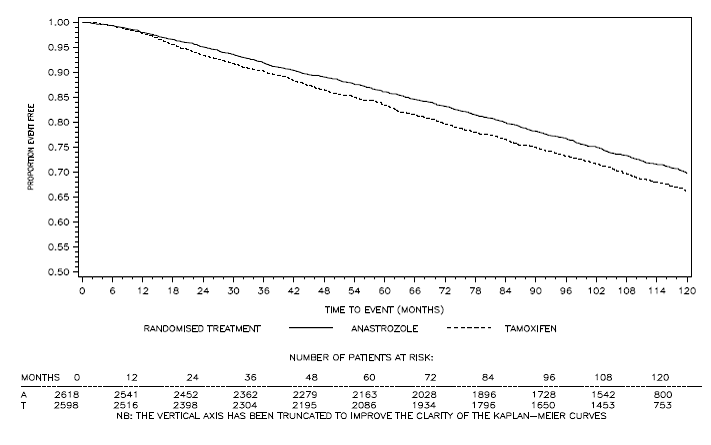
bPodíl pacientů se 120měsíčním sledováním byl 29,8 %.
Terapie první linie u žen po menopauze s pokročilým karcinomem prsu
Dvě dvojitě zaslepené, kontrolované klinické studie podobného designu (0030, severoamerická studie a 0027, převážně evropská studie) byly provedeny za účelem posouzení účinnosti přípravku ARIMIDEX 1 mg ve srovnání s tamoxifenem jako terapií první volby pro pozitivní hormonální receptory nebo hormonální receptory neznámý lokálně pokročilý nebo metastatický karcinom prsu u postmenopauzálních žen. Celkem 1021 pacientů ve věku od 30 do 92 let bylo randomizováno do zkušební léčby. Pacienti byli randomizováni k podávání 1 mg ARIMIDEXU 1 mg jednou denně nebo 20 mg tamoxifenu jednou denně. Primárními cílovými parametry pro obě studie byla doba do progrese nádoru, míra objektivní odpovědi nádoru a bezpečnost.
Demografické a další výchozí charakteristiky, včetně pacientů, kteří měli měřitelné a žádné měřitelné onemocnění, pacientů, kterým byla podávána předchozí adjuvantní léčba, místo metastatického onemocnění a etnický původ byly podobné pro obě léčebné skupiny v obou studiích. Následující tabulka shrnuje stav hormonálních receptorů při vstupu pro všechny randomizované pacientky ve studiích 0030 a 0027.
Pokud jde o primární cílové parametry, studie 0030 ukázala, že ARIMIDEX měl statisticky významnou výhodu oproti tamoxifenu (p=0,006) v době do progrese nádoru; míra objektivní odpovědi nádoru byla podobná pro ARIMIDEX 1 mg a tamoxifen. Studie 0027 ukázala, že ARIMIDEX 1 mg a tamoxifen měly podobnou objektivní míru odpovědi nádoru a dobu do progrese nádoru (viz tabulka 12 a obrázky 5 a 6).
Tabulka 12 níže shrnuje výsledky studie 0030 a studie 0027 pro primární cílové parametry účinnosti.
Obrázek 5: Kaplan-Meierova pravděpodobnost doby do progrese onemocnění u všech randomizovaných pacientů (intent-to-treat) ve studii 0030 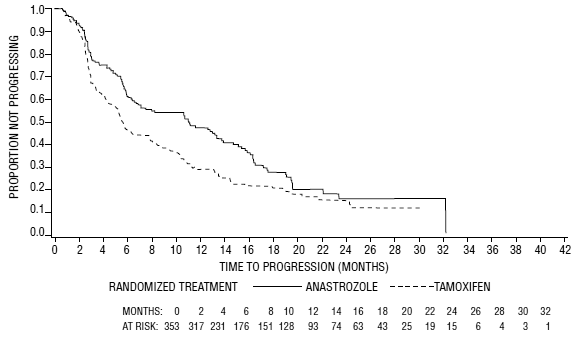
Obrázek 6: Kaplan-Meierova pravděpodobnost doby do progrese pro všechny randomizované pacienty (intent-to-treat) ve studii 0027 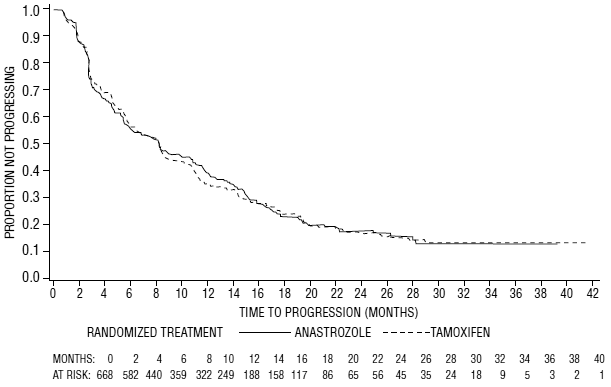
Výsledky ze sekundárních cílových bodů podpořily výsledky primárních cílových bodů účinnosti. Mezi léčebnými skupinami obou studií bylo příliš málo úmrtí, aby bylo možné vyvodit závěry o rozdílech v celkovém přežití.
Terapie druhé linie u žen po menopauze s pokročilou rakovinou prsu, které měly po léčbě tamoxifenem progresi onemocnění
Anastrozol byl zkoumán ve dvou kontrolovaných klinických studiích (0004, severoamerická studie; 0005, převážně evropská studie) u postmenopauzálních žen s pokročilým karcinomem prsu, u kterých došlo k progresi onemocnění po léčbě tamoxifenem pro pokročilý nebo časný karcinom prsu. Někteří z pacientů také podstoupili předchozí cytotoxickou léčbu. Většina pacientů byla ER-pozitivní; menší část byla ER-neznámá nebo ER-negativní; ER-negativní pacienti byli vhodní pouze v případě, že měli pozitivní odpověď na tamoxifen. Vhodní pacienti s měřitelným a neměřitelným onemocněním byli randomizováni tak, aby dostávali buď jednu denní dávku 1 mg nebo 10 mg ARIMIDEX nebo megestrol acetát 40 mg čtyřikrát denně. Studie byly dvojitě zaslepené s ohledem na ARIMIDEX. Primárními proměnnými účinnosti byla doba do progrese a míra objektivní odpovědi (pouze pacienti s měřitelnou chorobou mohli být považováni za částečné respondéry). Míry objektivních odpovědí byly vypočteny na základě kritérií Union Internationale Contre le Cancer (UICC). Byla také vypočtena míra prodloužené (více než 24 týdnů) stabilní nemoci, rychlost progrese a přežití.
Obě studie zahrnovaly více než 375 pacientů; demografické a další základní charakteristiky byly podobné pro tři léčebné skupiny v každé studii. Pacienti ve studii 0005 lépe reagovali na předchozí léčbu tamoxifenem. Ze zapsaných pacientek, které měly předchozí léčbu tamoxifenem pro pokročilé onemocnění (58 % ve studii 0004; 57 % ve studii 0005), 18 % těchto pacientek ve studii 0004 a 42 % ve studii 0005 uvedl primární zkoušející jako odpověď. Ve studii 0004 bylo 81 % pacientů ER-pozitivních, 13 % ER-neznámých a 6 % bylo ER-negativních. Ve studii 0005 bylo 58 % pacientů ER-pozitivních, 37 % ER-neznámých a 5 % bylo ER-negativních. Ve studii 0004 mělo měřitelné onemocnění 62 % pacientů ve srovnání se 79 % ve studii 0005. Místa metastatického onemocnění byla podobná mezi léčebnými skupinami v každé studii. V průměru mělo 40 % pacientů metastázy v měkkých tkáních; 60 % mělo kostní metastázy; a 40 % mělo viscerální (15 % jaterní) metastázy.
Výsledky účinnosti z těchto dvou studií byly podobné, jak je uvedeno v tabulce 13. V obou studiích nebyly žádné významné rozdíly mezi léčebnými rameny s ohledem na kterýkoli z parametrů účinnosti uvedených v tabulce níže.
Když se shromáždí údaje ze dvou kontrolovaných studií, míry objektivních odpovědí a medián časů do progrese a úmrtí byly podobné u pacientů randomizovaných na ARIMIDEX 1 mg a megestrol acetát. Tyto údaje nenaznačují, že ARIMIDEX 10 mg je lepší než ARIMIDEX 1 mg.
INFORMACE PRO PACIENTA
ARIMIDEX® Tablety A-rim-eh-dex (anastrozol) pro perorální podání
Jaké jsou nejdůležitější informace, které bych měl vědět o ARIMIDEXu?
ARIMIDEX může způsobit závažné nežádoucí účinky včetně:
- srdeční choroba. žen s časným karcinomem prsu, které mají v anamnéze ucpání srdečních tepen (ischemická choroba srdeční) a které užívají ARIMIDEX 1 mg, se mohou ve srovnání s podobnými ženami, které užívají tamoxifen, zvýšit příznaky sníženého průtoku krve do srdce.
Okamžitě vyhledejte lékařskou pomoc, pokud se u Vás během léčby přípravkem ARIMIDEX objeví nová nebo se zhorší bolest na hrudi nebo dušnost.
Co je ARIMIDEX?
ARIMIDEX 1 mg je lék na předpis používaný u žen po menopauze („změna života“) k:
- léčba časné rakoviny prsu
- po operaci
- u žen, jejichž rakovina prsu je pozitivní na hormonální receptory
- první léčba rakoviny prsu, která se rozšířila do blízké tkáně nebo lymfatických uzlin (lokálně pokročilé) nebo se rozšířila do jiných částí těla (metastatická), u žen, jejichž rakovina prsu je pozitivní na hormonální receptory nebo hormonální receptory nejsou známy
- léčba pokročilého karcinomu prsu, pokud se rakovina zvětšila nebo se onemocnění rozšířilo po léčbě tamoxifenem
ARIMIDEX 1 mg nepůsobí u žen s rakovinou prsu, které neprošly menopauzou (premenopauzální ženy).
Kdo by neměl užívat ARIMIDEX?
Neužívejte ARIMIDEX 1 mg, jestliže:
- jste těhotná nebo můžete otěhotnět. ARIMIDEX může poškodit vaše nenarozené dítě. Pokud otěhotníte během užívání přípravku ARIMIDEX, informujte ihned svého lékaře.
- neprošly menopauzou (jsou premenopauzální)
- měla závažnou alergickou reakci na anastrozol nebo na kteroukoli další složku přípravku ARIMIDEX. Úplný seznam složek přípravku ARIMIDEX naleznete na konci této příbalové informace. Příznaky závažné alergické reakce na ARIMIDEX zahrnují: otok obličeje, rtů, jazyka nebo hrdla, potíže s dýcháním nebo polykáním, kopřivku a svědění.
Co mám říci svému lékaři, než začnu užívat ARIMIDEX 1 mg?
Než začnete ARIMIDEX užívat, informujte svého lékaře, pokud:
- neprošly menopauzou. Pokud si nejste jistý(á), poraďte se se svým lékařem.
- máte nebo jste měli problémy se srdcem
- bylo vám řečeno, že máte řídnutí kostí nebo slabost (osteoporózu)
- mají vysoký cholesterol
- mít jiné zdravotní potíže
- jste těhotná nebo plánujete otěhotnět. ARIMIDEX může poškodit vaše nenarozené dítě. Viz "Kdo by neměl užívat ARIMIDEX?"
- kojíte nebo plánujete kojit. Není známo, zda ARIMIDEX přechází do mateřského mléka. Vy a váš lékař byste se měli rozhodnout, zda budete užívat ARIMIDEX 1 mg nebo kojit. Neměli byste dělat obojí.
Informujte svého lékaře o všech lécích, které užíváte, včetně léků na předpis a volně prodejných léků, vitamínů a bylinných doplňků. Zejména informujte svého lékaře, pokud užíváte:
- tamoxifen. Neměli byste brát ARIMIDEX jestliže užíváte tamoxifen. Užívání přípravku ARIMIDEX 1 mg s tamoxifenem může snížit množství přípravku ARIMIDEX 1 mg ve Vaší krvi a může způsobit, že přípravek ARIMIDEX 1 mg nebude také účinkovat.
- Léky, které obsahují estrogen. ARIMIDEX 1 mg nemusí účinkovat, pokud se užívá s některým z těchto léků:
- hormonální substituční terapie
- antikoncepční pilulky
- estrogenové krémy
- vaginální kroužky
- vaginální čípky
Znát léky, které užíváte. Uchovávejte si jejich seznam, abyste je mohli ukázat svému lékaři a lékárníkovi, když dostanete nový lék.
Jak mám užívat ARIMIDEX?
- Užívejte ARIMIDEX přesně podle pokynů svého lékaře.
- Pokračujte v užívání přípravku ARIMIDEX, dokud Vám lékař neřekne, abyste přestala.
- ARIMIDEX 1 mg lze užívat s jídlem nebo bez jídla.
- Pokud vynecháte dávku, užijte ji, jakmile si vzpomenete. Pokud je téměř čas na další dávku, vynechejte zapomenutou dávku. Vezměte si svou další pravidelnou dávku. Neužívejte dvě dávky současně.
Jestliže jste užil(a) příliš mnoho přípravku ARIMIDEX 1 mg, zavolejte svého lékaře nebo jděte ihned na pohotovost v nejbližší nemocnici.
Jaké jsou možné vedlejší účinky přípravku ARIMIDEX?
ARIMIDEX může způsobit závažné nežádoucí účinky včetně:
- Vidět "Jaké jsou nejdůležitější informace, které bych měl vědět o ARIMIDEX?"
- řídnutí nebo slabost kostí (osteoporóza). ARIMIDEX 1 mg snižuje estrogen ve vašem těle, což může způsobit řídnutí a oslabení vašich kostí. To může zvýšit riziko zlomenin, zejména páteře, kyčle a zápěstí. Váš lékař může nařídit test kostní minerální hustoty
- před zahájením léčby přípravkem ARIMIDEX a během ní, abyste si mohli zkontrolovat kostní změny.
- zvýšená hladina cholesterolu v krvi (tuk v krvi). Váš lékař může během užívání přípravku ARIMIDEX provést krevní testy, aby zkontroloval hladinu cholesterolu.
- kožní reakce. Přestaňte užívat ARIMIDEX 1 mg a ihned zavolejte svého lékaře, pokud se u Vás objeví jakékoli kožní léze, vředy nebo puchýře.
- závažné alergické reakce. Okamžitě vyhledejte lékařskou pomoc, pokud dostanete:
- otok obličeje, rtů, jazyka nebo hrdla
- potíže s polykáním nebo dýcháním
- problémy s játry. ARIMIDEX 1 mg může způsobit zánět jater a změny v krevních testech jaterních funkcí. Váš lékař vás může kvůli tomu zkontrolovat. Přestaňte užívat ARIMIDEX 1 mg a ihned kontaktujte svého lékaře jestliže máte některý z těchto známek nebo příznaků jaterního problému:
- obecný pocit, že není dobře
- zežloutnutí kůže nebo očního bělma
- bolest na pravé straně břicha (břicho)
Časté nežádoucí účinky u žen užívajících ARIMIDEX 1 mg zahrnují:
- návaly horka
- slabost
- bolesti kloubů
- bolest kloubů, ztuhlost nebo otoky (artritida)
- bolest
- bolest krku
- vysoký krevní tlak
- Deprese
- nevolnost a zvracení
- vyrážka
- bolesti zad
- problémy se spánkem
- bolest kostí
- bolest hlavy
- otoky nohou, kotníků nebo chodidel
- zvýšený kašel
- dušnost
- hromadění lymfatické tekutiny v tkáních postižené paže (lymfedém)
ARIMIDEX může také způsobit, že budete mít lechtání, brnění nebo necitlivost kůže.
Informujte svého lékaře, pokud máte jakýkoli nežádoucí účinek, který vás obtěžuje nebo který neustupuje.
Toto nejsou všechny možné vedlejší účinky přípravku ARIMIDEX. Pro více informací se zeptejte svého lékaře nebo lékárníka.
Zavolejte svého lékaře o radu ohledně nežádoucích účinků. Nežádoucí účinky můžete hlásit úřadu FDA na čísle 1-800-FDA-1088.
Jak mám uchovávat ARIMIDEX 1 mg?
- Uchovávejte ARIMIDEX 1 mg při pokojové teplotě mezi 68 °F až 77 °F (20 °C až 25 °C).
Uchovávejte ARIMIDEX a všechny léky mimo dosah dětí.
Obecné informace o bezpečném a účinném používání přípravku ARIMIDEX.
Léky jsou někdy předepisovány pro jiné účely, než které jsou uvedeny v příbalové informaci pro pacienty. Neužívejte ARIMIDEX 1 mg na onemocnění, pro které nebyl předepsán. Nepodávejte ARIMIDEX jiným lidem, i když mají stejné příznaky jako vy. Může jim to ublížit.
Pokud chcete více informací, promluvte si se svým lékařem. Můžete požádat svého lékárníka nebo lékaře o informace o přípravku ARIMIDEX, který je určen pro zdravotníky. Pro více informací volejte 1-866-992-9276 nebo navštivte www.ARIMIDEX.com.
Jaké jsou složky přípravku ARIMIDEX?
Aktivní složka: anastrozol
Neaktivní složky: laktóza, stearát hořečnatý, hydroxypropylmethylcelulóza, polyethylenglykol, povidon, sodná sůl glykolátu škrobu a oxid titaničitý.