Neurontin 100mg, 300mg, 400mg, 600mg Gabapentin Použití, vedlejší účinky a dávkování. Cena v internetové lékárně. Generické léky bez předpisu.
Co je Neurontin a jak se používá?
Neurontin 400 mg je lék na předpis používaný k léčbě příznaků bolesti nervů a záchvatů. Neurontin lze užívat samostatně nebo s jinými léky.
Neurontin je analog GABA, antiepileptikum.
Není známo, zda je Neurontin bezpečný a účinný u dětí mladších 3 let.
Jaké jsou možné vedlejší účinky Neurontinu 400 mg?
Neurontin může způsobit závažné nežádoucí účinky včetně:
- zvýšené záchvaty
- těžká slabost nebo únava
- problémy s rovnováhou nebo pohybem svalů
- bolest v horní části žaludku
- bolest na hrudi
- nový nebo zhoršující se kašel s horečkou
- potíže s dýcháním
- silné brnění nebo necitlivost
- rychlý pohyb očí
- malé nebo žádné močení
- bolestivé nebo obtížné močení
- otoky nohou nebo kotníků
Pokud máte některý z výše uvedených příznaků, okamžitě vyhledejte lékařskou pomoc.
Mezi nejčastější vedlejší účinky Neurontinu patří:
- bolest hlavy
- závrať
- ospalost
- únava
- otoky rukou nebo nohou
- problémy s očima
- problémy s koordinací
- Mezi nejčastější nežádoucí účinky přípravku Neurontin 600 mg u dětí patří:
- horečka
- nevolnost
- zvracení
- změny v chování
- problémy s pamětí
- potíže se soustředěním
- působí neklidně, nepřátelsky nebo agresivně
Informujte lékaře, pokud máte jakýkoli nežádoucí účinek, který vás obtěžuje nebo který neustupuje.
To nejsou všechny možné vedlejší účinky Neurontinu. Pro více informací se zeptejte svého lékaře nebo lékárníka.
Zavolejte svého lékaře o radu ohledně nežádoucích účinků. Nežádoucí účinky můžete hlásit úřadu FDA na čísle 1-800-FDA-1088.
POPIS
Účinnou látkou v kapslích, tabletách a perorálním roztoku NEURONTIN je gabapentin, který má chemický název kyselina 1-(aminomethyl)cyklohexanoctová.
Molekulární vzorec gabapentinu je C9H17NO2 a molekulová hmotnost je 171,24. Strukturní vzorec gabapentinu je:
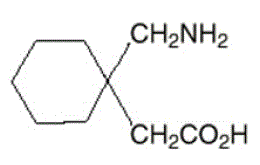
Gabapentin je bílá až téměř bílá krystalická pevná látka s pKal 3,7 a pKa2 10,7. Je volně rozpustný ve vodě a v zásaditých i kyselých vodných roztocích. Log rozdělovacího koeficientu (noctanol/0,05M fosfátový pufr) při pH 7,4 je –1,25.
Každá tobolka Neurontin 100 mg obsahuje 100 mg, 300 mg nebo 400 mg gabapentinu a následující neaktivní složky: laktózu, kukuřičný škrob, mastek, želatinu, oxid titaničitý, FD&C Blue č. 2, žlutý oxid železitý (pouze 300 mg a 400 mg) a červený oxid železitý (pouze 400 mg).
Každá tableta Neurontinu obsahuje 600 mg nebo 800 mg gabapentinu a následující neaktivní složky: poloxamer 407, kopovidon, kukuřičný škrob, stearát hořečnatý, hydroxypropylcelulóza, mastek a kandelilový vosk
Perorální roztok Neurontinu obsahuje 250 mg gabapentinu na 5 ml (50 mg na ml) a následující neaktivní složky: glycerin, xylitol, čištěnou vodu a umělou příchuť jahodového anýzu.
INDIKACE
NEURONTIN® je indikován pro:
- Léčba postherpetické neuralgie u dospělých
- Doplňková terapie při léčbě parciálních záchvatů, se sekundární generalizací a bez ní, u dospělých a dětských pacientů ve věku 3 let a starších s epilepsií
DÁVKOVÁNÍ A PODÁVÁNÍ
Dávkování pro postherpetickou neuralgii
U dospělých s postherpetickou neuralgií může být NEURONTIN zahájen 1. den v jedné dávce 300 mg, 2. den 600 mg/den (300 mg dvakrát denně) a 3. den 900 mg/den (300 mg 3x denně). krát denně). Dávku lze následně titrovat podle potřeby pro úlevu od bolesti na dávku 1800 mg/den (600 mg třikrát denně). V klinických studiích byla prokázána účinnost v rozmezí dávek od 1800 mg/den do 3600 mg/den se srovnatelnými účinky v celém dávkovém rozmezí; v těchto klinických studiích však nebyl prokázán další přínos použití dávek vyšších než 1800 mg/den.
Dávkování pro epilepsii s parciálními záchvaty
Pacienti ve věku 12 let a více
Počáteční dávka je 300 mg třikrát denně. Doporučená udržovací dávka přípravku NEURONTIN je 300 mg až 600 mg třikrát denně. V dlouhodobých klinických studiích byly dobře tolerovány dávky až do 2400 mg/den. Dávky 3600 mg/den byly také podávány malému počtu pacientů po relativně krátkou dobu a byly dobře tolerovány. Podávejte NEURONTIN třikrát denně pomocí 300 mg nebo 400 mg tobolek nebo 600 mg nebo 800 mg tablet. Maximální doba mezi dávkami by neměla přesáhnout 12 hodin.
Pediatričtí pacienti ve věku 3 až 11 let
Počáteční dávka je 10 mg/kg/den až 15 mg/kg/den, podaná ve třech dílčích dávkách, a doporučené udržovací dávky se dosáhne titrací směrem nahoru po dobu přibližně 3 dnů. Doporučená udržovací dávka přípravku NEURONTIN u pacientů ve věku 3 až 4 let je 40 mg/kg/den, podávaná ve třech dílčích dávkách. Doporučená udržovací dávka přípravku NEURONTIN 100 mg u pacientů ve věku 5 až 11 let je 25 mg/kg/den až 35 mg/kg/den, podávaná ve třech dílčích dávkách. NEURONTIN 300 mg může být podáván jako perorální roztok, kapsle nebo tableta nebo pomocí kombinací těchto přípravků. Dávky až do 50 mg/kg/den byly v dlouhodobé klinické studii dobře tolerovány. Maximální časový interval mezi dávkami by neměl přesáhnout 12 hodin.
Úprava dávkování u pacientů s poruchou funkce ledvin
Doporučuje se úprava dávkování u pacientů ve věku 12 let a starších s poruchou funkce ledvin nebo podstupujících hemodialýzu, a to následovně (viz doporučení ohledně dávkování výše pro účinné dávky v každé indikaci):
TABULKA 1: Dávkování NEURONTINU na základě funkce ledvin
Clearance kreatininu (CLCr) je u ambulantních pacientů obtížně měřitelná. U pacientů se stabilní funkcí ledvin lze clearance kreatininu přiměřeně dobře odhadnout pomocí rovnice Cockcrofta a Gaulta:
Použití přípravku NEURONTIN u pacientů mladších 12 let se sníženou funkcí ledvin nebylo studováno.
Dávkování u starších osob
Vzhledem k tomu, že u starších pacientů je pravděpodobnější, že budou mít sníženou funkci ledvin, je třeba věnovat pozornost výběru dávky a dávku upravit na základě hodnot clearance kreatininu u těchto pacientů.
Informace o administraci
Podávejte NEURONTIN perorálně s jídlem nebo bez jídla.
Tobolky NEURONTIN 100 mg se polykají celé a zapíjejí se vodou.
Informujte pacienty, že pokud si rozdělí 600mg nebo 800mg tabletu NEURONTIN s půlicí rýhou, aby si mohli podat polovinu tablety, měli by užít nepoužitou polovinu tablety jako další dávku. Půlky tablety nepoužité do 28 dnů od rozdělení tablety s půlicí rýhou je třeba zlikvidovat.
Pokud je dávka přípravku NEURONTIN snížena, vysazena nebo nahrazena alternativní léčbou, mělo by to být provedeno postupně po dobu minimálně 1 týdne (dle uvážení předepisujícího lékaře může být zapotřebí delší období).
JAK DODÁVÁNO
Dávkové formy A Síly
Kapsle
- 100 mg: bílé tvrdé želatinové tobolky s potiskem „PD“ na těle a „Neurontin/100 mg“ na víčku
- 300 mg: žluté tvrdé želatinové tobolky s potiskem „PD“ na těle a „Neurontin/300 mg“ na víčku
- 400 mg: oranžové tvrdé želatinové tobolky s potiskem „PD“ na těle a „Neurontin/400 mg“ na víčku
Tablety
- 600 mg: bílé eliptické potahované tablety s půlicí rýhou s vyraženým „NT“ a „16“ na jedné straně
- 800 mg: bílé eliptické potahované tablety s půlicí rýhou s vyraženým „NT“ a „26“ na jedné straně
Orální roztok
- 250 mg na 5 ml (50 mg na ml), čirý bezbarvý až slabě žlutý roztok
Skladování A Manipulace
NEURONTIN (gabapentin) tobolky, tablety a perorální roztok se dodávají následovně:
100 mg kapsle
Bílé tvrdé želatinové tobolky s potiskem „PD“ na těle a „Neurontin/100 mg“ na víčku; k dispozici v:
Láhve po 100: NDC 0071-0803-24
300 mg kapsle
Žluté tvrdé želatinové tobolky potištěné „PD“ na těle a „Neurontin/300 mg“ na víčku; k dispozici v:
Láhve po 100: NDC 0071-0805-24 Jednotková dávka 50: NDC 0071-0805-40
400 mg kapsle
Oranžové tvrdé želatinové tobolky s potiskem „PD“ na těle a „Neurontin/400 mg“ na víčku; k dispozici v:
Láhve po 100: NDC 0071-0806-24 Jednotková dávka 50: NDC 0071-0806-40
600 mg tablety
Bílé eliptické potahované tablety s půlicí rýhou s vyraženým „NT“ a „16“ na jedné straně; k dispozici v:
Láhve po 100: NDC 0071-0513-24
800 mg tablety
Bílé eliptické potahované tablety s půlicí rýhou s vyraženým „NT“ a „26“ na jedné straně; k dispozici v:
Láhve po 100: NDC 0071-0401-24
250 mg na 5 ml perorálního roztoku
Čirý bezbarvý až slabě žlutý roztok; každých 5 ml perorálního roztoku obsahuje 250 mg gabapentinu; k dispozici v:
Skleněné lahve o obsahu 470 ml: NDC 0071-2012-23 Lahvičky o obsahu 470 ml: NDC 0071-2012-44
Uchovávejte NEURONTIN 400 mg tablety a tobolky při teplotě 25 °C (77 °F); povolené odchylky mezi 15 °C až 30 °C (59 °F až 86 °F) [viz USP řízená pokojová teplota ].
Uchovávejte NEURONTIN perorální roztok v chladničce, 2 °C až 8 °C (36 °F až 46 °F).
Distribuce: Parke-Davis, divize společnosti Pfizer Inc., NY, NY 10017. Revize: prosinec 2020
VEDLEJŠÍ EFEKTY
Následující závažné nežádoucí účinky jsou podrobněji popsány v jiných částech:
- Léková reakce s eozinofilií a systémovými příznaky (DRESS) / Multiorgánová hypersenzitivita [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]
- Anafylaxe a angioedém [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]
- Somnolence/útlum a závratě [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]
- Abstinenční záchvat Sražený záchvat, Status Epilepticus [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]
- Sebevražedné chování a myšlenky [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]
- Respirační deprese [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]
- Neuropsychiatrické nežádoucí účinky (dětští pacienti ve věku 3 až 12 let) [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]
- Náhlá a nevysvětlitelná smrt u pacientů s epilepsií [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]
Zkušenosti z klinických studií
Vzhledem k tomu, že klinické studie jsou prováděny za velmi odlišných podmínek, nelze míry nežádoucích reakcí pozorované v klinických studiích léku přímo srovnávat s mírami v klinických studiích jiného léku a nemusí odrážet míry pozorované v praxi.
Postherpetická neuralgie
Nejčastějšími nežádoucími účinky spojenými s užíváním přípravku NEURONTIN u dospělých, které nebyly pozorovány s ekvivalentní frekvencí u pacientů léčených placebem, byly závratě, somnolence a periferní edém.
Ve 2 kontrolovaných studiích u postherpetické neuralgie přerušilo léčbu kvůli nežádoucí reakci 16 % z 336 pacientů, kteří dostávali NEURONTIN, a 9 % z 227 pacientů, kteří dostávali placebo. Nežádoucí účinky, které nejčastěji vedly k vysazení u pacientů léčených přípravkem NEURONTIN, byly závratě, ospalost a nauzea.
Tabulka 3 uvádí nežádoucí účinky, které se vyskytly u nejméně 1 % pacientů léčených přípravkem NEURONTIN s postherpetickou neuralgií účastnících se placebem kontrolovaných studií a které byly číselně častější ve skupině s přípravkem NEURONTIN než ve skupině s placebem.
TABULKA 3: Nežádoucí reakce ve sdružených placebem kontrolovaných studiích u postherpetické neuralgie
Další reakce u více než 1 % pacientů, ale stejně nebo častěji ve skupině s placebem, zahrnovaly bolest, třes, neuralgii, bolest zad, dyspepsii, dušnost a chřipkový syndrom.
Mezi muži a ženami nebyly žádné klinicky významné rozdíly v typech a výskytu nežádoucích účinků. Protože bylo málo pacientů, jejichž rasa byla hlášena jako jiná než bílá, nejsou k dispozici dostatečné údaje, které by podpořily tvrzení týkající se rozložení nežádoucích účinků podle rasy.
Epilepsie s parciálními záchvaty (přídavná léčba)
Nejčastějšími nežádoucími účinky přípravku NEURONTIN 300 mg v kombinaci s jinými antiepileptiky u pacientů ve věku >12 let, které nebyly pozorovány s ekvivalentní frekvencí u pacientů léčených placebem, byly ospalost, závratě, ataxie, únava a nystagmus.
Nejčastějšími nežádoucími účinky přípravku NEURONTIN v kombinaci s jinými antiepileptiky u pediatrických pacientů ve věku 3 až 12 let, které nebyly pozorovány se stejnou frekvencí u pacientů léčených placebem, byly virová infekce, horečka, nauzea a/nebo zvracení, ospalost a nepřátelství [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Přibližně 7 % z 2 074 pacientů ve věku >12 let a přibližně 7 % ze 449 pediatrických pacientů ve věku 3 až 12 let, kteří dostávali NEURONTIN v klinických studiích před uvedením na trh, přerušilo léčbu kvůli nežádoucí reakci. Nežádoucí účinky nejčastěji spojené s vysazením pacientů ve věku >12 let byly ospalost (1,2 %), ataxie (0,8 %), únava (0,6 %), nauzea a/nebo zvracení (0,6 %) a závratě (0,6 %) . Nežádoucí reakce nejčastěji spojené s vysazením u pediatrických pacientů byly emoční labilita (1,6 %), hostilita (1,3 %) a hyperkineze (1,1 %).
Tabulka 4 uvádí nežádoucí účinky, které se vyskytly u nejméně 1 % pacientů léčených přípravkem NEURONTIN ve věku >12 let s epilepsií, kteří se účastnili placebem kontrolovaných studií a byly číselně častější ve skupině léčené přípravkem NEURONTIN. V těchto studiích byl k pacientově současné léčbě antiepileptiky přidán buď NEURONTIN 300 mg nebo placebo.
TABULKA 4: Nežádoucí reakce ve sdružených placebem kontrolovaných přídavných studiích u pacientů s epilepsií ve věku >12 let
Mezi nežádoucími účinky vyskytujícími se u pacientů léčených přípravkem NEURONTIN s incidencí alespoň 10 % se zdálo, že ospalost a ataxie vykazují pozitivní vztah mezi dávkou a odezvou.
Celkový výskyt nežádoucích účinků a typy pozorovaných nežádoucích účinků byly u mužů a žen léčených přípravkem NEURONTIN podobné. Výskyt nežádoucích účinků se mírně zvyšoval s rostoucím věkem u pacientů léčených buď přípravkem NEURONTIN nebo placebem. Protože pouze 3 % pacientů (28/921) v placebem kontrolovaných studiích bylo identifikováno jako nebílí (černí nebo jiní), nejsou k dispozici dostatečné údaje, které by podpořily tvrzení týkající se rozdělení nežádoucích účinků podle rasy.
Tabulka 5 uvádí nežádoucí účinky, které se vyskytly u nejméně 2 % pacientů léčených přípravkem NEURONTIN ve věku 3 až 12 let s epilepsií účastnících se placebem kontrolovaných studií a které byly číselně častější ve skupině léčené přípravkem NEURONTIN.
TABULKA 5: Nežádoucí reakce v placebem kontrolované přídavné studii u pacientů s dětskou epilepsií ve věku 3 až 12 let
Další reakce u více než 2 % pediatrických pacientů ve věku 3 až 12 let, ale stejně nebo častěji ve skupině s placebem, zahrnovaly: faryngitidu, infekci horních cest dýchacích, bolest hlavy, rýmu, křeče, průjem, anorexii, kašel a zánět středního ucha.
Postmarketingové zkušenosti
Během postmarketingového používání přípravku NEURONTIN byly zjištěny následující nežádoucí účinky. Protože jsou tyto reakce hlášeny dobrovolně z populace nejisté velikosti, není vždy možné spolehlivě odhadnout jejich frekvenci nebo stanovit příčinnou souvislost s expozicí léku.
Poruchy jater a žlučových cest: žloutenka
Vyšetřování: zvýšená kreatinkináza, zvýšené jaterní testy
Poruchy metabolismu a výživy: hyponatrémie
Poruchy svalové a kosterní soustavy a pojivové tkáně: rabdomyolýza
Poruchy nervového systému: pohybová porucha
Psychiatrické poruchy: míchání
Poruchy reprodukčního systému a prsou: zvětšení prsou, změny libida, poruchy ejakulace a anorgasmie
Poruchy kůže a podkoží: angioedém [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ], bulózní pemfigoid, erythema multiforme, Stevens-Johnsonův syndrom.
Existují postmarketingové zprávy o život ohrožující nebo fatální respirační depresi u pacientů užívajících NEURONTIN s opioidy nebo jinými látkami tlumícími CNS nebo při základní respirační poruše (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Byly také hlášeny nežádoucí účinky po náhlém vysazení gabapentinu. Nejčastěji hlášenými reakcemi byly úzkost, nespavost, nauzea, bolest a pocení.
DROGOVÉ INTERAKCE
Opioidy
Po současném podání gabapentinu s opioidy (např. morfinem, hydrokodonem, oxykodonem, buprenorfinem) byly hlášeny respirační deprese a sedace, někdy vedoucí k úmrtí. VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Hydrokodon
Současné podávání přípravku NEURONTIN 300 mg s hydrokodonem snižuje expozici hydrokodonu [viz KLINICKÁ FARMAKOLOGIE ]. Při zahájení nebo ukončení léčby přípravkem NEURONTIN 100 mg u pacienta užívajícího hydrokodon je třeba vzít v úvahu možnost změny expozice a účinku hydrokodonu.
Morfium
Když je gabapentin podáván s morfinem, měli by být pacienti sledováni kvůli známkám deprese CNS, jako je somnolence, sedace a respirační deprese (viz KLINICKÁ FARMAKOLOGIE ].
Jiná antiepileptika
Gabapentin není znatelně metabolizován ani neinterferuje s metabolismem běžně současně podávaných antiepileptik (viz KLINICKÁ FARMAKOLOGIE ].
Maalox® (hydroxid hlinitý, hydroxid hořečnatý)
Průměrná biologická dostupnost gabapentinu byla snížena přibližně o 20 % při současném užívání antacid (Maalox) obsahujících hydroxidy hořčíku a hliníku. Doporučuje se užívat gabapentin alespoň 2 hodiny po podání Maaloxu [viz KLINICKÁ FARMAKOLOGIE ].
Interakce drog/laboratorních testů
Protože byly hlášeny falešně pozitivní výsledky testu Ames N-Multistix SG pro stanovení bílkovin v moči, když byl gabapentin přidán k jiným antiepileptikům, doporučuje se ke stanovení přítomnosti bílkovin v moči specifičtější postup srážení kyselinou sulfosalicylovou.
Zneužívání drog a závislost
Kontrolovaná látka
Gabapentin není plánovaný lék.
Zneužívání
Zneužívání je záměrné, neterapeutické použití drogy, byť jednorázové, pro její žádoucí psychologické nebo fyziologické účinky. Zneužití je úmyslné užití drogy k terapeutickým účelům jednotlivcem jiným způsobem, než je předepsáno poskytovatelem zdravotní péče nebo komu nebylo předepsáno.
Gabapentin nevykazuje afinitu k benzodiazepinovým, opioidním (mu, delta nebo kappa) nebo kanabinoidním 1 receptorovým místům. Zneužívání a zneužívání gabapentinu bylo hlášeno po uvedení přípravku na trh a v publikované literatuře. Většina jedinců popsaných v těchto zprávách měla v anamnéze zneužívání více látek. Někteří z těchto jedinců užívali vyšší než doporučené dávky gabapentinu pro neschválená použití. Při předepisování přípravku NEURONTIN pečlivě vyhodnoťte u pacientů anamnézu zneužívání drog a sledujte, zda se u nich nevyskytují známky a příznaky nesprávného užívání nebo zneužívání gabapentinu (např. eskalace vlastní dávky a vyhledávání drog). Potenciál zneužívání gabapentinu nebyl ve studiích na lidech hodnocen.
Závislost
Fyzická závislost je stav, který vzniká v důsledku fyziologické adaptace v reakci na opakované užívání drogy, projevující se abstinenčními příznaky a symptomy po náhlém vysazení nebo výrazném snížení dávky drogy. Existují vzácné postmarketingové zprávy o jedincích, u kterých se krátce po ukončení užívání vyšších než doporučených dávek gabapentinu používaného k léčbě onemocnění, pro něž není tento lék schválen, objevily abstinenční příznaky. Tyto symptomy zahrnovaly agitovanost, dezorientaci a zmatenost po náhlém vysazení gabapentinu, které vymizely po opětovném zahájení léčby gabapentinem. Potenciál závislosti na gabapentinu nebyl hodnocen ve studiích na lidech.
VAROVÁNÍ
Zahrnuto jako součást OPATŘENÍ sekce.
OPATŘENÍ
Léková reakce s eozinofilií a systémovými příznaky (DRESS)/Multiorgánová hypersenzitivita
přípravku NEURONTIN se objevila léková reakce s eozinofilií a systémovými příznaky (DRESS), také známá jako multiorgánová hypersenzitivita. Některé z těchto reakcí byly fatální nebo život ohrožující. DRESS se typicky, i když ne výlučně, projevuje horečkou, vyrážkou a/nebo lymfadenopatií ve spojení s postižením jiného orgánového systému, jako je hepatitida, nefritida, hematologické abnormality, myokarditida nebo myositida, někdy připomínající akutní virovou infekci. Často je přítomna eozinofilie. Tato porucha je variabilní ve svém projevu a mohou se na ní podílet další orgánové systémy, které zde nejsou uvedeny.
Je důležité si uvědomit, že časné projevy přecitlivělosti, jako je horečka nebo lymfadenopatie, mohou být přítomny, i když vyrážka není zjevná. Pokud jsou takové známky nebo příznaky přítomny, pacient by měl být okamžitě vyšetřen. Přípravek NEURONTIN by měl být vysazen, pokud nelze zjistit alternativní etiologii známek nebo symptomů.
Anafylaxe A Angioedém
NEURONTIN může způsobit anafylaxi a angioedém po první dávce nebo kdykoli během léčby. Známky a příznaky v hlášených případech zahrnovaly potíže s dýcháním, otok rtů, hrdla a jazyka a hypotenzi vyžadující pohotovostní léčbu. Pacienti by měli být poučeni, aby přerušili léčbu přípravkem NEURONTIN 300 mg a okamžitě vyhledali lékařskou pomoc, pokud se u nich objeví známky nebo příznaky anafylaxe nebo angioedému.
Účinky na řízení a provoz těžkých strojů
Pacienti užívající NEURONTIN by neměli řídit, dokud nezískají dostatečné zkušenosti k posouzení, zda NEURONTIN 100 mg zhoršuje jejich schopnost řídit. Studie jízdního výkonu provedené s prolékem gabapentinu (tableta gabapentinu enacarbil, s prodlouženým uvolňováním) ukazují, že gabapentin může způsobit významné zhoršení řízení. Předepisující lékaři a pacienti by si měli být vědomi toho, že pacienti' schopnost posoudit své vlastní řidičské schopnosti, stejně jako jejich schopnost posoudit stupeň ospalosti způsobené přípravkem NEURONTIN, může být nedokonalá. Doba trvání poruchy řízení po zahájení léčby přípravkem NEURONTIN 300 mg není známa. Zda postižení souvisí s ospalostí [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ] nebo jiné účinky přípravku NEURONTIN nejsou známy.
Navíc, protože NEURONTIN 300 mg způsobuje somnolenci a závratě [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ], je třeba pacientům doporučit, aby neobsluhovali složité stroje, dokud nezískají dostatečné zkušenosti s přípravkem NEURONTIN, aby bylo možné posoudit, zda přípravek NEURONTIN 300 mg zhoršuje jejich schopnost vykonávat takové úkoly.
Ospalost/sedace a závratě
Během kontrolovaných studií epilepsie u pacientů starších 12 let, kteří dostávali dávky 100 mg až 1 800 mg NEURONTIN denně, byla ospalost, závratě a ataxie hlášeny ve vyšší míře u pacientů užívajících NEURONTIN ve srovnání s placebem: tj. 19 % v léku. oproti 9 % u placeba pro somnolenci, 17 % u léku oproti 7 % u placeba pro závratě a 13 % u léku oproti 6 % u placeba pro ataxii. V těchto studiích byly ospalost, ataxie a únava častými nežádoucími účinky, které vedly k přerušení léčby přípravkem NEURONTIN u pacientů starších 12 let, přičemž 1,2 %, 0,8 % a 0,6 % přerušilo léčbu pro tyto příhody.
Během kontrolovaných studií u pacientů s postherpetickou neuralgií byla ospalost a závratě hlášeny ve vyšší míře ve srovnání s placebem u pacientů užívajících NEURONTIN v dávkách až 3600 mg denně: tj. 21 % u pacientů léčených přípravkem NEURONTIN oproti 5 % u pacientů léčených placebem pro somnolenci a 28 % u pacientů léčených přípravkem NEURONTIN oproti 8 % u pacientů léčených placebem pro závratě. Závratě a somnolence patřily k nejčastějším nežádoucím účinkům vedoucím k přerušení léčby přípravkem NEURONTIN.
Pokud je přípravek NEURONTIN 400 mg užíván s jinými léky se sedativními vlastnostmi, je třeba u pacientů pečlivě sledovat příznaky deprese centrálního nervového systému (CNS), jako je ospalost a sedace, a to z důvodu možné synergie. Navíc u pacientů, kteří vyžadují současnou léčbu morfinem, může dojít ke zvýšení koncentrací gabapentinu a může vyžadovat úpravu dávky (viz DROGOVÉ INTERAKCE ].
Abstinenční záchvat Sražený záchvat, Status Epilepticus
Antiepileptika by neměla být náhle vysazena kvůli možnosti zvýšené frekvence záchvatů.
V placebem kontrolovaných studiích epilepsie u pacientů starších 12 let byla incidence status epilepticus u pacientů užívajících NEURONTIN 0,6 % (3 z 543) oproti 0,5 % u pacientů užívajících placebo (2 z 378). Z 2074 pacientů ve věku >12 let léčených přípravkem NEURONTIN 300 mg ve všech studiích epilepsie (kontrolované a nekontrolované) mělo 31 (1,5 %) status epilepticus. Z toho 14 pacientů nemělo v anamnéze status epilepticus ani před léčbou, ani během užívání jiných léků. Vzhledem k tomu, že nejsou k dispozici adekvátní historické údaje, není možné říci, zda je léčba přípravkem NEURONTIN spojena s vyšší nebo nižší četností status epilepticus, než by se dalo očekávat u podobné populace, která nebyla léčena přípravkem NEURONTIN.
Sebevražedné Chování A Nápady
Antiepileptika (AED), včetně přípravku NEURONTIN 100 mg, zvyšují riziko sebevražedných myšlenek nebo chování u pacientů užívajících tyto léky z jakékoli indikace. Pacienti léčení jakýmkoliv AED pro jakoukoli indikaci by měli být sledováni z hlediska výskytu nebo zhoršení deprese, sebevražedných myšlenek nebo chování a/nebo jakýchkoli neobvyklých změn nálady nebo chování.
Souhrnné analýzy 199 placebem kontrolovaných klinických studií (mono- a přídatná terapie) s 11 různými AED ukázaly, že pacienti randomizovaní k jednomu z AED měli přibližně dvojnásobné riziko (upravené relativní riziko 1,8, 95% CI:1,2, 2,7) sebevraždy myšlení nebo chování ve srovnání s pacienty randomizovanými k placebu. V těchto studiích, které měly medián trvání léčby 12 týdnů, byla odhadovaná míra výskytu sebevražedného chování nebo myšlenek u 27 863 pacientů léčených AED 0,43 % ve srovnání s 0,24 % u 16 029 pacientů léčených placebem, což představuje nárůst přibližně o jeden případ sebevražedného myšlení nebo chování na každých 530 léčených pacientů. Ve studiích došlo ke čtyřem sebevraždám u pacientů léčených drogami a k žádné u pacientů léčených placebem, ale toto číslo je příliš malé na to, aby bylo možné učinit závěr o účinku léku na sebevraždu.
Zvýšené riziko sebevražedných myšlenek nebo chování u AED bylo pozorováno již týden po zahájení medikamentózní léčby AED a přetrvávalo po celou dobu hodnocené léčby. Protože většina studií zahrnutých do analýzy nepřesáhla 24 týdnů, riziko sebevražedných myšlenek nebo chování po 24 týdnech nebylo možné posoudit.
Riziko sebevražedných myšlenek nebo chování bylo u drog v analyzovaných datech obecně konzistentní. Zjištění zvýšeného rizika u AED s různým mechanismem účinku a napříč řadou indikací naznačuje, že riziko se týká všech AED používaných pro jakoukoli indikaci. V analyzovaných klinických studiích se riziko podstatně nelišilo podle věku (5 – 100 let). Tabulka 2 ukazuje absolutní a relativní riziko podle indikace pro všechna hodnocená AED.
TABULKA 2: Riziko podle indikace pro antiepileptika v souhrnné analýze
Relativní riziko sebevražedných myšlenek nebo chování bylo vyšší v klinických studiích pro epilepsii než v klinických studiích pro psychiatrické nebo jiné stavy, ale rozdíly v absolutním riziku byly podobné pro epilepsii a psychiatrické indikace.
Každý, kdo zvažuje předepsání přípravku NEURONTIN 100 mg nebo jakéhokoli jiného AED, musí vyvážit riziko sebevražedných myšlenek nebo chování s rizikem neléčené nemoci. Epilepsie a mnoho dalších nemocí, pro které se předepisují AED, jsou samy o sobě spojeny s nemocností a úmrtností a zvýšeným rizikem sebevražedných myšlenek a chování. Pokud se během léčby objeví sebevražedné myšlenky a chování, musí předepisující lékař zvážit, zda výskyt těchto příznaků u některého pacienta může souviset s léčeným onemocněním.
Pacienti, jejich pečovatelé a rodiny by měli být informováni, že AED zvyšují riziko sebevražedných myšlenek a chování, a měli by být upozorněni na nutnost dávat pozor na výskyt nebo zhoršení známek a příznaků deprese, jakékoli neobvyklé změny nálady nebo chování. , nebo vznik sebevražedných myšlenek, chování nebo myšlenek na sebepoškozování. Chování vzbuzující obavy by mělo být okamžitě hlášeno poskytovatelům zdravotní péče.
Respirační deprese
Existují důkazy z kazuistik, studií na lidech a studií na zvířatech, které spojují gabapentin se závažnou, život ohrožující nebo fatální respirační depresí při současném podávání s látkami tlumícími CNS, včetně opioidů, nebo v případě základní respirační poruchy. Pokud se rozhodnete předepsat NEURONTIN 100 mg společně s jiným tlumivým prostředkem na CNS, zejména opioidem, nebo předepsat NEURONTIN 100 mg pacientům se základní respirační poruchou, sledujte pacienty, zda se u nich nevyskytují příznaky respirační deprese a sedace, a zvažte zahájení léčby přípravkem NEURONTIN v nízké dávce. . Léčba deprese dýchání může zahrnovat pečlivé sledování, podpůrná opatření a snížení nebo vysazení látek tlumících CNS (včetně NEURONTINu).
Neuropsychiatrické nežádoucí účinky (dětští pacienti ve věku 3 až 12 let)
Užívání gabapentinu u pediatrických pacientů s epilepsií ve věku 3 až 12 let je spojeno s výskytem nežádoucích účinků souvisejících s CNS. Nejvýznamnější z nich lze rozdělit do následujících kategorií: 1) emoční labilita (především problémy s chováním), 2) hostilita, včetně agresivního chování, 3) porucha myšlení, včetně problémů s koncentrací a změny školního prospěchu, a 4) hyperkineze ( především neklid a hyperaktivita). U pacientů léčených gabapentinem byla většina reakcí mírné až střední intenzity.
kontrolovaných klinických studiích s epilepsií u pediatrických pacientů ve věku 3 až 12 let byla incidence těchto nežádoucích účinků: emoční labilita 6 % (pacienti léčení gabapentinem) versus 1,3 % (pacienti léčení placebem); nepřátelství 5,2 % versus 1,3 %; hyperkineze 4,7 % versus 2,9 %; a porucha myšlení 1,7 % oproti 0 %. Jedna z těchto reakcí, zpráva o nepřátelství, byla považována za vážnou. K přerušení léčby gabapentinem došlo u 1,3 % pacientů uvádějících emoční labilitu a hyperkinezi au 0,9 % pacientů léčených gabapentinem uvádějících hostilitu a poruchu myšlení. Jeden pacient léčený placebem (0,4 %) odstoupil z důvodu emoční lability.
Tumorogenní potenciál
Ve studii kancerogenity po perorálním podání gabapentin zvýšil výskyt nádorů pankreatických acinárních buněk u potkanů (viz Neklinická toxikologie ]. Klinický význam tohoto nálezu není znám. Klinické zkušenosti během premarketingového vývoje gabapentinu neposkytují žádné přímé prostředky k posouzení jeho potenciálu pro indukci nádorů u lidí.
V klinických studiích adjuvantní terapie u epilepsie zahrnujících 2 085 pacientoroků expozice u pacientů >12 let byly nové nádory hlášeny u 10 pacientů (2 prsu, 3 mozkové, 2 plíce, 1 nadledvinka, 1 non-Hodgkin' s lymfomem, 1 karcinomem endometria in situ) a již existujícími nádory se zhoršily u 11 pacientek (9 mozku, 1 prsu, 1 prostaty) během nebo až 2 roky po vysazení přípravku NEURONTIN. Bez znalosti základního výskytu a recidivy u podobné populace, která nebyla léčena přípravkem NEURONTIN 100 mg, není možné vědět, zda výskyt pozorovaný v této kohortě je či není ovlivněn léčbou.
Náhlá a nevysvětlitelná smrt u pacientů s epilepsií
průběhu předmarketingového vývoje přípravku NEURONTIN 400 mg bylo zaznamenáno 8 náhlých a nevysvětlitelných úmrtí u kohorty 2203 pacientů s epilepsií léčených (2103 pacientoroků expozice) přípravkem NEURONTIN.
Některé z nich by mohly představovat úmrtí související se záchvaty, kdy záchvat nebyl pozorován, např. v noci. To představuje výskyt 0,0038 úmrtí na pacientorok. Ačkoli tato míra převyšuje míru očekávanou u zdravé populace odpovídající věku a pohlaví, je v rozmezí odhadů výskytu náhlých nevysvětlitelných úmrtí u pacientů s epilepsií, kteří nedostávají NEURONTIN (v rozmezí od 0,0005 pro běžnou populaci epileptiků do 0,003 pro populaci v klinické studii podobnou té v programu NEURONTIN, na 0,005 u pacientů s refrakterní epilepsií). V důsledku toho, zda jsou tato čísla uklidňující nebo vyvolávají další obavy, závisí na srovnatelnosti populací hlášených v kohortě NEURONTIN a přesnosti poskytnutých odhadů.
Informace pro pacienty
Doporučte pacientovi, aby si přečetl označení pacienta schválené FDA ( Průvodce léky ).
Informace o administraci
Informujte pacienty, že NEURONTIN se užívá perorálně s jídlem nebo bez jídla. Informujte pacienty, že pokud si rozdělí 600mg nebo 800mg tabletu s půlicí rýhou, aby si mohli podat polovinu tablety, měli by užít nepoužitou polovinu tablety jako další dávku. Informujte pacienty, aby zlikvidovali nepoužité poloviny tablet do 28 dnů od rozdělení tablety s půlicí rýhou.
Léková reakce s eozinofilií a systémovými příznaky (DRESS)/Multiorgánová hypersenzitivita
Před zahájením léčby přípravkem NEURONTIN poučte pacienty, že vyrážka nebo jiné známky nebo příznaky přecitlivělosti (jako je horečka nebo lymfadenopatie) mohou být předzvěstí závažné zdravotní příhody a že pacient by měl jakýkoli takový výskyt okamžitě hlásit lékaři [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Anafylaxe A Angioedém
Informujte pacienty, aby přerušili léčbu přípravkem NEURONTIN 300 mg a vyhledali lékařskou péči, pokud se u nich objeví známky nebo příznaky anafylaxe nebo angioedému (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Závratě a ospalost a účinky na řízení a obsluhu těžkých strojů
Informujte pacienty, že přípravek NEURONTIN 100 mg může způsobit závratě, ospalost a další příznaky a známky deprese CNS. Jiné léky se sedativními vlastnostmi mohou tyto příznaky zvýšit. V souladu s tím, ačkoli pacienti' schopnost určit úroveň jejich postižení může být nespolehlivá, doporučte jim, aby neřídili auto ani neobsluhovali jiné složité stroje, dokud nezískají dostatečné zkušenosti s přípravkem NEURONTIN, aby mohli posoudit, zda nepříznivě ovlivňuje jejich duševní a/nebo motorický výkon. Informujte pacienty, že není známo, jak dlouho tento účinek trvá [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ a VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Sebevražedné myšlení a chování
Informujte pacienta, jeho pečovatele a rodiny, že AED, včetně NEURONTINu, mohou zvýšit riziko sebevražedných myšlenek a chování. Informujte pacienty o nutnosti být ostražití při vzniku nebo zhoršení příznaků deprese, jakýchkoli neobvyklých změn nálady nebo chování nebo výskytu sebevražedných myšlenek, chování nebo myšlenek na sebepoškozování. Poučte pacienty, aby chování, které je znepokojuje, okamžitě hlásili poskytovatelům zdravotní péče [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Respirační deprese
Informujte pacienty o riziku respirační deprese. Zahrňte informaci, že riziko je největší u těch, kteří současně užívají léky tlumící CNS (jako jsou opioidní analgetika) nebo u těch, kteří mají základní respirační poruchu. Naučte pacienty, jak rozpoznat respirační depresi, a doporučte jim, aby okamžitě vyhledali lékařskou pomoc, pokud k ní dojde [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Použití v těhotenství
Poučte pacienty, aby informovaly svého lékaře, pokud otěhotní nebo plánují otěhotnět během léčby, a aby informovaly svého lékaře, pokud kojí nebo plánují kojit během léčby [viz Použití u konkrétních populací ].
Vyzvěte pacientky, aby se zapsaly do registru těhotenství NAAED, pokud otěhotní. Tento registr shromažďuje informace o bezpečnosti antiepileptik během těhotenství. Pro registraci mohou pacienti zavolat na bezplatné číslo 1-888-233-2334 [viz Použití u konkrétních populací ].
Označení tohoto produktu mohlo být aktualizováno. Nejnovější informace o předepisování naleznete na www.pfizer.com.
Neklinická toxikologie
Karcinogeneze, mutageneze, zhoršení plodnosti
Karcinogeneze
Gabapentin byl podáván perorálně myším a potkanům ve dvouletých studiích karcinogenity. U myší léčených dávkami až 2000 mg/kg/den nebyl pozorován žádný důkaz karcinogenity související s léčivem. Při dávce 2000 mg/kg byla plazmatická expozice gabapentinu (AUC) u myší přibližně 2krát vyšší než u lidí při MRHD 3600 mg/den. U potkanů bylo u samců potkanů, kteří dostávali nejvyšší dávku (2000 mg/kg), ale ne v dávkách 250 nebo 1000 mg/kg/den, zjištěno zvýšení výskytu adenomu acinárních buněk pankreatu a karcinomu. Při dávce 1000 mg/kg byla plazmatická expozice gabapentinu (AUC) u potkanů přibližně 5krát vyšší než u lidí při MRHD.
Studie navržené ke zkoumání mechanismu gabapentinem indukované pankreatické karcinogeneze u potkanů ukazují, že gabapentin stimuluje syntézu DNA v potkaních pankreatických acinárních buňkách in vitro, a proto může působit jako nádorový promotor zvýšením mitogenní aktivity. Není známo, zda má gabapentin schopnost zvyšovat buněčnou proliferaci u jiných typů buněk nebo u jiných druhů, včetně lidí.
Mutageneze
Gabapentin nevykazoval mutagenní ani genotoxický potenciál in vitro (Amesův test, HGPRT test dopředné mutace v plicních buňkách čínského křečka) a in vivo (chromozomální aberace a mikronukleární test v kostní dřeni čínského křečka, myší mikronukleus, neplánovaná syntéza DNA v potkaních hepatocytech) testy.
Zhoršení Plodnosti
U potkanů při dávkách do 2000 mg/kg nebyly pozorovány žádné nežádoucí účinky na fertilitu nebo reprodukci. Při dávce 2000 mg/kg je plazmatická expozice gabapentinu (AUC) u potkanů přibližně 8krát vyšší než u lidí při MRHD.
Použití u konkrétních populací
Těhotenství
Registr expozice v těhotenství
Existuje registr těhotenských expozic, který sleduje výsledky těhotenství u žen vystavených během těhotenství antiepileptikům (AED), jako je NEURONTIN 300 mg. Povzbuďte ženy, které během těhotenství užívají přípravek NEURONTIN, aby se zapsaly do registru těhotenství severoamerických antiepileptických léků (NAAED) zavoláním na bezplatné číslo 1-888-233-2334 nebo na http://www.aedpregnancyregistry.org/.
Shrnutí rizik
Nejsou k dispozici dostatečné údaje o vývojových rizicích spojených s užíváním přípravku NEURONTIN u těhotných žen. V neklinických studiích na myších, potkanech a králících byl gabapentin vývojově toxický (zvýšené fetální skeletální a viscerální abnormality a zvýšená embryofetální mortalita), když byl podáván březím zvířatům v dávkách podobných nebo nižších, než jaké se používají klinicky [viz Data ].
obecné populaci USA je odhadované základní riziko závažných vrozených vad a potratu u klinicky uznaných těhotenství 2 až 4 % a 15 až 20 %. Základní riziko závažných vrozených vad a potratu u uvedené populace není známo.

Data
Údaje o zvířatech
Když březí myši dostávaly perorální dávky gabapentinu (500, 1000 nebo 3000 mg/kg/den) během období organogeneze, byla pozorována embryofetální toxicita (zvýšený výskyt kosterních variací) při dvou nejvyšších dávkách. Dávka bez účinku na embryofetální vývojovou toxicitu u myší (500 mg/kg/den) je nižší než maximální doporučená dávka pro člověka (MRHD) 3600 mg na základě plochy povrchu těla (mg/m²).
Ve studiích, ve kterých potkany dostávaly perorální dávky gabapentinu (500 až 2000 mg/kg/den) během březosti, byly pozorovány nežádoucí účinky na vývoj potomstva (zvýšený výskyt hydroureteru a/nebo hydronefrózy) při všech dávkách. Nejnižší testovaná dávka je podobná MRHD na základě mg/m².
Když byly březí králice léčeny gabapentinem během období organogeneze, bylo pozorováno zvýšení embryofetální mortality ve všech testovaných dávkách (60, 300 nebo 1500 mg/kg). Nejnižší testovaná dávka je nižší než MRHD na základě mg/m².
V publikované studii byl gabapentin (400 mg/kg/den) podáván intraperitoneální injekcí neonatálním myším během prvního postnatálního týdne, období synaptogeneze u hlodavců (odpovídající poslednímu trimestru těhotenství u lidí). Gabapentin způsobil výrazné snížení tvorby neuronálních synapsí v mozcích intaktních myší a abnormální tvorbu neuronálních synapsí v myším modelu synaptické opravy. Bylo prokázáno, že gabapentin in vitro interferuje s aktivitou α2δ podjednotky napěťově aktivovaných kalciových kanálů, receptoru zapojeného do neuronální synaptogeneze. Klinický význam těchto nálezů není znám.
Laktace
Shrnutí rizik
Gabapentin se po perorálním podání vylučuje do mateřského mléka. Účinky na kojené dítě a na produkci mléka nejsou známy. Vývojové a zdravotní přínosy kojení by měly být zváženy spolu s klinickou potřebou matky NEURONTIN a veškerými potenciálními nežádoucími účinky NEURONTINu nebo základního stavu matky na kojené dítě.
Pediatrické použití
Bezpečnost a účinnost přípravku NEURONTIN 600 mg v léčbě postherpetické neuralgie u pediatrických pacientů nebyla stanovena.
Bezpečnost a účinnost jako doplňkové terapie při léčbě parciálních záchvatů u pediatrických pacientů mladších 3 let nebyla stanovena (viz Klinické studie ].
Geriatrické použití
Celkový počet pacientů léčených přípravkem NEURONTIN v kontrolovaných klinických studiích u pacientů s postherpetickou neuralgií byl 336, z toho 102 (30 %) bylo ve věku 65 až 74 let a 168 (50 %) bylo ve věku 75 let a více. U pacientů ve věku 75 let a starších byl větší účinek léčby ve srovnání s mladšími pacienty, kteří dostávali stejnou dávku. Vzhledem k tomu, že gabapentin je téměř výhradně eliminován renální exkrecí, větší účinek léčby pozorovaný u pacientů ve věku ≥ 75 let může být důsledkem zvýšené expozice gabapentinu při dané dávce, která je důsledkem poklesu renálních funkcí souvisejícím s věkem. Nelze však vyloučit ani další faktory. Typy a incidence nežádoucích účinků byly ve všech věkových skupinách podobné, s výjimkou periferního edému a ataxie, které měly tendenci se s věkem zvyšovat.
Klinické studie přípravku NEURONTIN u epilepsie nezahrnovaly dostatečný počet subjektů ve věku 65 a více let, aby bylo možné určit, zda reagovali odlišně od mladších subjektů. Jiné hlášené klinické zkušenosti nezjistily rozdíly v odpovědích mezi staršími a mladšími pacienty. Obecně by měl být výběr dávky pro staršího pacienta opatrný, obvykle začínající na spodní hranici dávkovacího rozmezí, což odráží vyšší frekvenci snížené funkce jater, ledvin nebo srdce a souběžného onemocnění nebo jiné lékové terapie.
Je známo, že tento lék je v podstatě vylučován ledvinami a riziko toxických reakcí na tento lék může být větší u pacientů s poruchou funkce ledvin. Vzhledem k tomu, že u starších pacientů je pravděpodobnější, že budou mít sníženou funkci ledvin, je třeba věnovat pozornost výběru dávky a dávka by měla být upravena na základě hodnot clearance kreatininu u těchto pacientů (viz DÁVKOVÁNÍ A PODÁVÁNÍ , NEŽÁDOUCÍ REAKCE , a KLINICKÁ FARMAKOLOGIE ].
Renální poškození
Úprava dávkování u dospělých pacientů se zhoršenou funkcí ledvin je nezbytná (viz DÁVKOVÁNÍ A PODÁVÁNÍ a KLINICKÁ FARMAKOLOGIE ]. Pediatričtí pacienti s renální insuficiencí nebyli studováni.
Úprava dávkování u pacientů podstupujících hemodialýzu je nezbytná [viz DÁVKOVÁNÍ A PODÁVÁNÍ a KLINICKÁ FARMAKOLOGIE ].
PŘEDÁVKOVAT
Příznaky akutní toxicity u zvířat zahrnovaly ataxii, ztížené dýchání, ptózu, sedaci, hypoaktivitu nebo excitaci.
Bylo hlášeno akutní předávkování perorálním přípravkem NEURONTIN 300 mg. Příznaky zahrnovaly dvojité vidění, třes, nezřetelnou řeč, ospalost, změněný duševní stav, závratě, letargii a průjem. Fatální respirační deprese byla hlášena při předávkování přípravkem NEURONTIN 400 mg, samotným nebo v kombinaci s jinými látkami tlumícími CNS.
Gabapentin lze odstranit hemodialýzou.
Pokud dojde k přeexponování, zavolejte do toxikologického centra na číslo 1-800-222-1222.
KONTRAINDIKACE
NEURONTIN je kontraindikován u pacientů, u kterých byla prokázána přecitlivělost na léčivo nebo jeho složky.
KLINICKÁ FARMAKOLOGIE
Mechanismus působení
Přesné mechanismy, kterými gabapentin vyvolává své analgetické a antiepileptické účinky, nejsou známy. Gabapentin je strukturně příbuzný neurotransmiteru gama-aminomáselné kyselině (GABA), ale nemá žádný vliv na vazbu, vychytávání nebo degradaci GABA. Studie in vitro ukázaly, že gabapentin se s vysokou afinitou váže na podjednotku α2δ napěťově aktivovaných vápníkových kanálů; vztah této vazby k terapeutickým účinkům gabapentinu však není znám.
Farmakokinetika
Všechny farmakologické účinky po podání gabapentinu jsou způsobeny aktivitou mateřské sloučeniny; gabapentin není u lidí znatelně metabolizován.
Orální biologická dostupnost
Biologická dostupnost gabapentinu není úměrná dávce; tj. jak se dávka zvyšuje, biologická dostupnost klesá. Biologická dostupnost gabapentinu je přibližně 60 %, 47 %, 34 %, 33 % a 27 % po dávkách 900, 1200, 2400, 3600 a 4800 mg/den podaných ve 3 dílčích dávkách. Jídlo má pouze malý vliv na rychlost a rozsah absorpce gabapentinu (14% zvýšení AUC a Cmax).
Rozdělení
Méně než 3 % gabapentinu cirkuluje vázaného na plazmatické bílkoviny. Zdánlivý distribuční objem gabapentinu po intravenózním podání 150 mg je 58±6 l (průměr ±SD). U pacientů s epilepsií byly koncentrace gabapentinu v cerebrospinálním moku v ustáleném stavu před dávkou (Cmin) přibližně 20 % odpovídajících plazmatických koncentrací.
Odstranění
Gabapentin je eliminován ze systémové cirkulace renální exkrecí jako nezměněný lék. Gabapentin není u lidí znatelně metabolizován.
Poločas eliminace gabapentinu je 5 až 7 hodin a nemění se dávkou ani po opakovaných dávkách. Konstanta rychlosti eliminace gabapentinu, plazmatická clearance a renální clearance jsou přímo úměrné clearance kreatininu. U starších pacientů au pacientů s poruchou funkce ledvin je plazmatická clearance gabapentinu snížena. Gabapentin lze z plazmy odstranit hemodialýzou.
Specifické populace
Stáří
Vliv věku byl studován u jedinců ve věku 20–80 let. Zdánlivá perorální clearance (CL/F) gabapentinu se s rostoucím věkem snižovala, z přibližně 225 ml/min u osob mladších 30 let na přibližně 125 ml/min u osob starších 70 let. Renální clearance (CLr) a CLr upravené podle tělesného povrchu také klesaly s věkem; nicméně pokles renální clearance gabapentinu s věkem lze z velké části vysvětlit poklesem renálních funkcí. [vidět DÁVKOVÁNÍ A PODÁVÁNÍ a Použití u konkrétních populací ].
Rod
Ačkoli nebyla provedena žádná formální studie, která by porovnávala farmakokinetiku gabapentinu u mužů a žen, zdá se, že farmakokinetické parametry pro muže a ženy jsou podobné a neexistují žádné významné rozdíly mezi pohlavími.
Závod
Farmakokinetické rozdíly způsobené rasou nebyly studovány. Vzhledem k tomu, že gabapentin je primárně vylučován ledvinami a neexistují žádné významné rasové rozdíly v clearance kreatininu, neočekávají se farmakokinetické rozdíly způsobené rasou.
Pediatrická
Farmakokinetika gabapentinu byla stanovena u 48 pediatrických pacientů ve věku od 1 měsíce do 12 let po dávce přibližně 10 mg/kg. Maximální plazmatické koncentrace byly podobné v celé věkové skupině a objevily se 2 až 3 hodiny po podání dávky. Obecně platí, že pediatričtí jedinci ve věku od 1 měsíce do
Populační farmakokinetická analýza byla provedena u 253 pediatrických pacientů ve věku od 1 měsíce do 13 let. Pacienti dostávali 10 až 65 mg/kg/den podávaných třikrát denně. Zdánlivá perorální clearance (CL/F) byla přímo úměrná clearance kreatininu a tento vztah byl podobný po jedné dávce a v ustáleném stavu. Vyšší hodnoty perorální clearance byly pozorovány u dětí ve věku
Tyto farmakokinetické údaje naznačují, že účinná denní dávka u pediatrických pacientů s epilepsií ve věku 3 a 4 let by měla být 40 mg/kg/den, aby se dosáhlo průměrných plazmatických koncentrací podobných těm, které se dosahují u pacientů ve věku 5 let a starších užívajících gabapentin v dávce 30 mg/den kg/den [viz DÁVKOVÁNÍ A PODÁVÁNÍ ].
Dospělí pacienti s poruchou ledvin
Subjektům (N=60) s poruchou funkce ledvin (průměrná clearance kreatininu v rozmezí 13–114 ml/min) byla podávána jednorázová perorální dávka 400 mg gabapentinu. Průměrný poločas gabapentinu se pohyboval od přibližně 6,5 hodiny (pacienti s clearance kreatininu >60 ml/min) do 52 hodin (clearance kreatininu 60 ml/min skupina) na přibližně 10 ml/min ( DÁVKOVÁNÍ A PODÁVÁNÍ a Použití u konkrétních populací ]. Pediatričtí pacienti s renální insuficiencí nebyli studováni.
Hemodialýza
Ve studii s anurickými dospělými subjekty (N=11) byl zdánlivý poločas eliminace gabapentinu ve dnech bez dialýzy asi 132 hodin; během dialýzy se zdánlivý poločas gabapentinu zkrátil na 3,8 hodiny. Hemodialýza má tedy významný vliv na eliminaci gabapentinu u anurických subjektů [viz DÁVKOVÁNÍ A PODÁVÁNÍ a Použití u konkrétních populací ].
Onemocnění jater
Vzhledem k tomu, že gabapentin není metabolizován, nebyla provedena žádná studie u pacientů s poruchou funkce jater.
Lékové interakce
Studie in vitro
Byly provedeny studie in vitro s cílem prozkoumat potenciál gabapentinu inhibovat hlavní enzymy cytochromu P450 (CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1 a CYP3A4), které zprostředkovávají metabolismus léčiv a xenobiotik pomocí izoformně selektivních markerových substrátů a lidských jaterních mikrozomálních přípravků . Pouze při nejvyšší testované koncentraci (171 mcg/ml; 1 mM) byl pozorován mírný stupeň inhibice (14 % až 30 %) izoformy CYP2A6. Žádná inhibice žádné z ostatních testovaných izoforem nebyla pozorována při koncentracích gabapentinu až do 171 mcg/ml (přibližně 15násobek C při 3600 mg/den) pozorované izoformy CYP2A6. Žádná inhibice žádné z dalších testovaných izoforem nebyla pozorována při koncentracích gabapentinu až do 171 mcg/ml (přibližně 15násobek Cmax při 3600 mg/den).
Studie in vivo
Údaje o lékových interakcích popsané v této části byly získány ze studií zahrnujících zdravé dospělé a dospělé pacienty s epilepsií.
fenytoin
Ve studii s jednorázovou (400 mg) a opakovanou dávkou (400 mg třikrát denně) přípravku NEURONTIN 400 mg u pacientů s epilepsií (N=8), kteří byli na monoterapii fenytoinem po dobu nejméně 2 měsíců, neměl gabapentin žádný účinek na minimální rovnovážný stav plazmatické koncentrace fenytoinu a fenytoinu neměly žádný vliv na farmakokinetiku gabapentinu.
karbamazepin
Koncentrace karbamazepinu a karbamazepinu 10, 11 v rovnovážném stavu nebyly ovlivněny současným podáváním gabapentinu (400 mg třikrát denně; N=12). Podobně farmakokinetika gabapentinu nebyla podáváním karbamazepinu změněna.
kyselina valproová
Průměrné sérové koncentrace kyseliny valproové v ustáleném stavu před a během současného podávání gabapentinu (400 mg třikrát denně; N=17) se nelišily a ani farmakokinetické parametry gabapentinu nebyly ovlivněny kyselinou valproovou.
fenobarbital
Odhady farmakokinetických parametrů v ustáleném stavu pro fenobarbital nebo gabapentin (300 mg třikrát denně; N=12) jsou identické bez ohledu na to, zda jsou léky podávány samostatně nebo společně.
naproxen
Zdá se, že současné podávání (N=18) tobolek sodné soli naproxenu (250 mg) s NEURONTINEM (125 mg) zvyšuje množství absorbovaného gabapentinu o 12 % až 15 %. Gabapentin neměl žádný vliv na farmakokinetické parametry naproxenu. Tyto dávky jsou nižší než terapeutické dávky pro obě léčiva. Rozsah interakce v rámci doporučených dávek obou léků není znám.
Hydrokodon
Současné podávání NEURONTINU (125 až 500 mg; N=48) snižuje hodnoty Cmax a AUC hydrokodonu (10 mg; N=50) v závislosti na dávce ve srovnání s podáváním samotného hydrokodonu; Hodnoty Cmax a AUC jsou o 3 % až 4 % nižší po podání 125 mg NEURONTINU a o 21 % až 22 % nižší po podání 500 mg NEURONTINU. Mechanismus této interakce není znám. Hydrokodon zvyšuje hodnoty AUC gabapentinu o 14 %. Velikost interakce při jiných dávkách není známa.
Morfium
Literární článek uvádí, že když byla podána 60mg tobolka morfinu s řízeným uvolňováním 2 hodiny před tobolkou 600mg NEURONTIN (N=12), průměrná AUC gabapentinu se zvýšila o 44 % ve srovnání s gabapentinem podávaným bez morfinu. Hodnoty farmakokinetických parametrů morfinu nebyly podáním NEURONTINU 2 hodiny po morfinu ovlivněny. Velikost interakce při jiných dávkách není známa.
Cimetidin
přítomnosti cimetidinu v dávce 300 mg čtyřikrát denně (N=12) klesla průměrná zdánlivá perorální clearance gabapentinu o 14 % a clearance kreatininu klesla o 10 %. Zdá se tedy, že cimetidin mění renální vylučování gabapentinu i kreatininu, endogenního markeru renálních funkcí. Neočekává se, že by toto malé snížení vylučování gabapentinu cimetidinem mělo klinický význam. Účinek gabapentinu na cimetidin nebyl hodnocen.
Orální antikoncepce
Na základě AUC a poločasu byly farmakokinetické profily více dávek norethindronu a ethinylestradiolu po podání tablet obsahujících 2,5 mg norethindron acetátu a 50 mcg ethinylestradiolu podobné při současném podávání gabapentinu a bez něj (400 mg třikrát denně; N=13). Cmax norethindronu byla o 13 % vyšší, když byl podáván společně s gabapentinem; neočekává se, že by tato interakce měla klinický význam.
Antacida (Maalox®) (hydroxid hlinitý, hydroxid hořečnatý)
Antacida (Maalox®) obsahující hydroxidy hořčíku a hliníku snížily průměrnou biologickou dostupnost gabapentinu (N=16) asi o 20 %. Toto snížení biologické dostupnosti bylo asi 10 %, když byl gabapentin podán 2 hodiny po Maaloxu.
Probenecid
Probenecid je blokátor renální tubulární sekrece. Farmakokinetické parametry gabapentinu bez a s probenecidem byly srovnatelné. To naznačuje, že gabapentin nepodléhá renální tubulární sekreci cestou, která je blokována probenecidem.
Klinické studie
Postherpetická neuralgie
NEURONTIN byl hodnocen pro léčbu postherpetické neuralgie (PHN) ve dvou randomizovaných, dvojitě zaslepených, placebem kontrolovaných, multicentrických studiích. Populaci intent-to-treat (ITT) tvořilo celkem 563 pacientů s bolestí po dobu delší než 3 měsíce po zhojení kožní vyrážky herpes zoster (tabulka 6).
TABULKA 6. Kontrolované studie PHN: trvání, dávky a počet pacientů
Každá studie zahrnovala 7- nebo 8-týdenní dvojitě zaslepenou fázi (3 nebo 4 týdny titrace a 4 týdny fixní dávky). Pacienti zahájili léčbu titrací na maximální dávku 900 mg/den gabapentinu po dobu 3 dnů. Dávky pak měly být titrovány v přírůstcích 600 až 1200 mg/den ve 3- až 7denních intervalech na cílovou dávku během 3 až 4 týdnů. Pacienti zaznamenávali svou bolest do denního deníku pomocí 11bodové číselné stupnice hodnocení bolesti v rozsahu od 0 (žádná bolest) do 10 (nejhorší možná bolest). Pro randomizaci bylo vyžadováno průměrné skóre bolesti během výchozího stavu alespoň 4. Analýzy byly provedeny s použitím populace ITT (všichni randomizovaní pacienti, kteří dostali alespoň jednu dávku studovaného léku).
Obě studie prokázaly účinnost ve srovnání s placebem ve všech testovaných dávkách.
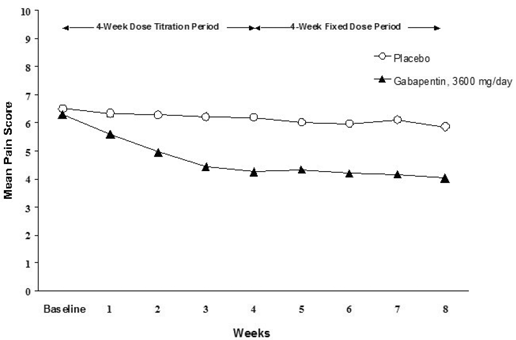
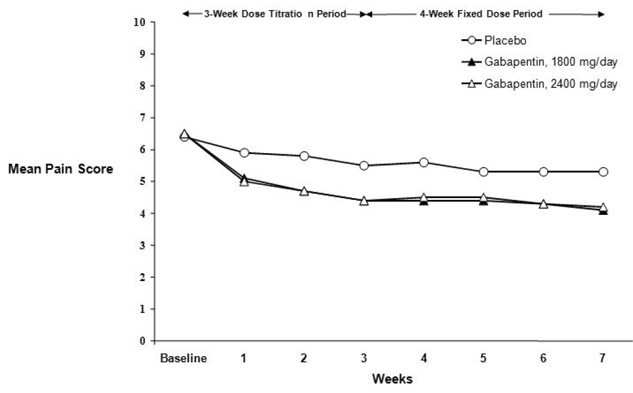
Snížení průměrného týdenního skóre bolesti bylo pozorováno v 1. týdnu v obou studiích a bylo zachováno až do konce léčby. Srovnatelné léčebné účinky byly pozorovány ve všech ramenech aktivní léčby. Farmakokinetické/farmakodynamické modelování poskytlo potvrzující důkaz účinnosti napříč všemi dávkami. Obrázky 1 a 2 ukazují skóre intenzity bolesti v průběhu času pro studie 1 a 2.
Obrázek 1. Týdenní průměrné skóre bolesti (pozorované případy v populaci ITT): Studie 1
Obrázek 2. Týdenní průměrné skóre bolesti (pozorované případy v populaci ITT): Studie 2
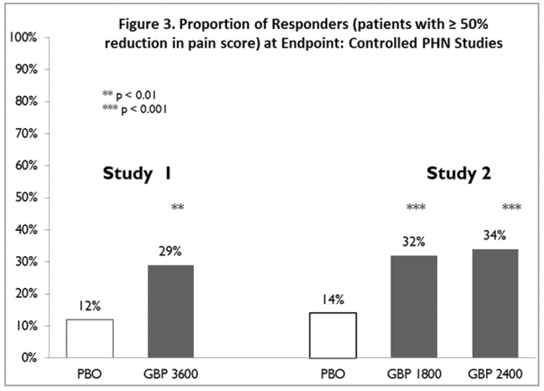
Pro každou studii byl vypočítán podíl respondentů (pacienti, kteří uváděli alespoň 50% zlepšení koncového skóre bolesti ve srovnání s výchozí hodnotou) (obrázek 3).
Obrázek 3. Podíl respondentů (pacienti s ≥50% snížením skóre bolesti) v koncovém bodě: kontrolované studie PHN
Epilepsie pro parciální záchvaty (přídavná léčba)
Účinnost přípravku NEURONTIN 400 mg jako doplňkové léčby (přidané k jiným antiepileptikům) byla stanovena v multicentrických placebem kontrolovaných, dvojitě zaslepených klinických studiích s paralelními skupinami u dospělých a dětských pacientů (3 roky a starší) s refrakterními parciálními záchvaty.
Důkazy účinnosti byly získány ve třech studiích provedených u 705 pacientů (ve věku 12 let a více) a jedné studii provedené u 247 pediatrických pacientů (ve věku 3 až 12 let). Zařazení pacienti měli v anamnéze alespoň 4 parciální záchvaty za měsíc navzdory tomu, že dostávali jedno nebo více antiepileptik v terapeutických hladinách a byli pozorováni na jejich zavedeném antiepileptickém léčebném režimu během 12týdenního základního období (6 týdnů ve studii pediatrické pacientů). U pacientů, u kterých se nadále vyskytovaly alespoň 2 (nebo 4 v některých studiích) záchvaty za měsíc, byl poté ke stávající léčbě během 12týdenního léčebného období přidán přípravek NEURONTIN 300 mg nebo placebo. Účinnost byla hodnocena především na základě procenta pacientů s 50% nebo větším snížením frekvence záchvatů od výchozího stavu k léčbě ("míra odpovědí") a odvozené míry nazývané poměr odpovědí, míra změny definovaná jako (T - B)/(T + B), kde B je výchozí frekvence záchvatů pacienta a T je frekvence záchvatů pacienta během léčby. Poměr odezvy je distribuován v rozsahu -1 až +1. Nulová hodnota znamená žádnou změnu, zatímco úplné odstranění záchvatů by dalo hodnotu -1; zvýšená míra záchvatů by poskytla kladné hodnoty. Poměr odezvy -0,33 odpovídá 50% snížení frekvence záchvatů. Níže uvedené výsledky jsou pro všechny parciální záchvaty v populaci intent-to-treat (všichni pacienti, kteří dostali jakékoli dávky léčby) v každé studii, pokud není uvedeno jinak.
Jedna studie porovnávala přípravek NEURONTIN 1200 mg/den ve třech dílčích dávkách s placebem. Míra odpovědí byla 23 % (14/61) ve skupině NEURONTIN 600 mg a 9 % (6/66) ve skupině s placebem; rozdíl mezi skupinami byl statisticky významný. Poměr odpovědí byl také lepší ve skupině NEURONTIN (-0,199) než ve skupině s placebem (-0,044), což je rozdíl, který rovněž dosáhl statistické významnosti.
Druhá studie porovnávala primárně NEURONTIN 1200 mg/den ve třech dílčích dávkách (N=101) s placebem (N=98). Další menší skupiny dávkování NEURONTIN 300 mg (600 mg/den, N=53; 1800 mg/den, N=54) byly také studovány pro informace týkající se odpovědi na dávku. Míra odpovědí byla vyšší ve skupině s přípravkem NEURONTIN 1200 mg/den (16 %) než ve skupině s placebem (8 %), ale rozdíl nebyl statisticky významný. Míra respondérů na 600 mg (17 %) také nebyla významně vyšší než u placeba, ale míra respondérů ve skupině s 1800 mg (26 %) byla statisticky významně vyšší než u placeba. Poměr odpovědí byl lepší ve skupině s přípravkem NEURONTIN 1200 mg/den (-0,103) než ve skupině s placebem (-0,022); ale tento rozdíl také nebyl statisticky významný (p = 0,224). Lepší odpověď byla pozorována ve skupině NEURONTIN 600 mg/den (-0,105) a 1800 mg/den (-0,222) než ve skupině 1200 mg/den, přičemž skupina 1800 mg/den dosáhla statistické významnosti ve srovnání s placebem skupina.
Třetí studie porovnávala přípravek NEURONTIN 900 mg/den ve třech dílčích dávkách (N=111) a placebo (N=109). Další dávková skupina NEURONTIN 1200 mg/den (N=52) poskytla údaje o reakci na dávku. Statisticky významný rozdíl v četnosti odpovědí byl pozorován ve skupině s přípravkem NEURONTIN 900 mg/den (22 %) ve srovnání se skupinou s placebem (10 %). Poměr odpovědí byl také statisticky významně lepší ve skupině NEURONTIN 900 mg/den (-0,119) ve srovnání se skupinou s placebem (-0,027), stejně jako poměr odpovědí ve skupině NEURONTIN 1200 mg/den (-0,184) ve srovnání s placebem.
každé studii byly také provedeny analýzy ke zkoumání účinku přípravku NEURONTIN 300 mg na prevenci sekundárně generalizovaných tonicko-klonických záchvatů. Do těchto analýz byli zahrnuti pacienti, kteří ve všech třech placebem kontrolovaných studiích prodělali sekundárně generalizovaný tonicko-klonický záchvat buď ve výchozím stavu, nebo v období léčby. Bylo provedeno několik srovnání poměru odpovědí, která ukázala statisticky významnou výhodu pro NEURONTIN ve srovnání s placebem a příznivé trendy pro téměř všechna srovnání.
Analýza míry respondérů s použitím kombinovaných dat ze všech tří studií a všech dávek (N=162, NEURONTIN; N=89, placebo) také ukázala významnou výhodu NEURONTINu oproti placebu ve snížení frekvence sekundárně generalizovaných tonicko-klonických záchvatů.
Ve dvou ze tří kontrolovaných studií byla použita více než jedna dávka přípravku NEURONTIN. V rámci každé studie výsledky neprokázaly konzistentně zvýšenou odpověď na dávku. Při pohledu napříč studiemi je však zřejmý trend ke zvyšování účinnosti se zvyšující se dávkou (viz obrázek 4).
Obrázek 4. Míra odpovědí u pacientů užívajících NEURONTIN 600 mg vyjádřená jako rozdíl od placeba podle dávky a studie: Studie doplňkové terapie u pacientů ve věku ≥ 12 let s parciálními záchvaty
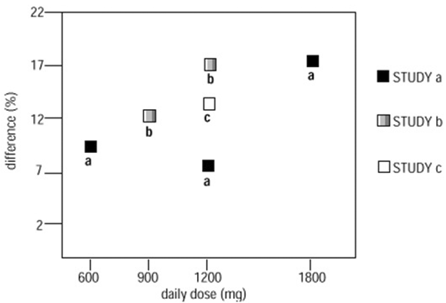
Na obrázku je vynesena velikost účinku léčby, měřená na ose Y jako rozdíl v podílu pacientů s gabapentinem a placebem, kteří dosáhli 50% nebo většího snížení frekvence záchvatů oproti výchozí hodnotě, proti denní dávce podávaného gabapentinu. (osa X).
Ačkoli nebyla provedena žádná formální analýza podle pohlaví, odhady odpovědi (poměr odpovědi) odvozené z klinických studií (398 mužů, 307 žen) naznačují, že neexistují žádné významné rozdíly mezi pohlavími. Neexistoval žádný konzistentní vzorec naznačující, že věk měl nějaký vliv na odpověď na NEURONTIN. Nebyl dostatečný počet pacientů jiné rasy než kavkazské, aby bylo možné porovnat účinnost mezi rasovými skupinami.
Čtvrtá studie u pediatrických pacientů ve věku 3 až 12 let porovnávala 25–35 mg/kg/den NEURONTIN (N=118) s placebem (N=127). U všech parciálních záchvatů v populaci intent-to-treat byl poměr odpovědí statisticky významně lepší pro skupinu NEURONTIN (-0,146) než pro skupinu s placebem (-0,079). U stejné populace se míra odpovědí na NEURONTIN (21 %) významně nelišila od placeba (18 %).
Studie u pediatrických pacientů ve věku 1 měsíc až 3 roky srovnávala 40 mg/kg/den NEURONTIN (N=38) s placebem (N=38) u pacientů, kteří dostávali alespoň jedno antiepileptikum na trhu a měli alespoň jeden parciální záchvat během období screeningu (do 2 týdnů před výchozí hodnotou). Pacienti měli až 48 hodin výchozího stavu a až 72 hodin dvojitě zaslepeného video EEG monitorování pro záznam a počítání výskytu záchvatů. Nebyly žádné statisticky významné rozdíly mezi léčbami ani v poměru odezvy, ani v míře respondérů.
INFORMACE PRO PACIENTA
NEURONTIN (Neu ron' tin) (Gabapentin) Kapsle, tablety a perorální roztok
Jaké jsou nejdůležitější informace, které bych měl vědět o přípravku NEURONTIN?
Nepřestávejte užívat přípravek NEURONTIN 300 mg, aniž byste se nejprve poradili se svým poskytovatelem zdravotní péče.
Náhlé zastavení přípravku NEURONTIN může způsobit vážné problémy.
NEURONTIN 400 mg může způsobit závažné nežádoucí účinky včetně:
1. Sebevražedné myšlenky. Stejně jako ostatní antiepileptika může NEURONTIN 100 mg vyvolat sebevražedné myšlenky nebo činy u velmi malého počtu lidí, asi u 1 z 500.
Okamžitě zavolejte poskytovatele zdravotní péče, pokud máte některý z těchto příznaků, zejména pokud jsou nové, horší nebo vás znepokojují:
- myšlenky na sebevraždu nebo smrt
- pokusy o sebevraždu
- nová nebo horší deprese
- nová nebo horší úzkost
- pocit vzrušení nebo neklidu
- panický záchvat
- potíže se spánkem (nespavost)
- nová nebo horší podrážděnost
- chovat se agresivně, být naštvaný nebo násilný
- působící na nebezpečné impulsy
- extrémní nárůst aktivity a mluvení (mánie)
- jiné neobvyklé změny v chování nebo náladě
Jak mohu sledovat časné příznaky sebevražedných myšlenek a činů?
- Věnujte pozornost jakýmkoli změnám, zejména náhlým změnám nálady, chování, myšlenek nebo pocitů.
- Udržujte všechny následné návštěvy u svého poskytovatele zdravotní péče podle plánu.
Zavolejte svého poskytovatele zdravotní péče mezi návštěvami podle potřeby, zvláště pokud máte obavy z příznaků.
Nepřestávejte užívat přípravek NEURONTIN, aniž byste se nejprve poradili s poskytovatelem zdravotní péče.
- Náhlé vysazení přípravku NEURONTIN 100 mg může způsobit vážné problémy. Náhlé vysazení léku na záchvaty u pacienta s epilepsií může způsobit záchvaty, které nepřestanou (status epilepticus).
- Sebevražedné myšlenky nebo činy mohou být způsobeny jinými věcmi než léky. Pokud máte sebevražedné myšlenky nebo činy, váš poskytovatel zdravotní péče může zkontrolovat jiné příčiny.
2. Změny v chování a myšlení - Užívání přípravku NEURONTIN 100 mg u dětí od 3 do 12 let může způsobit emoční změny, agresivní chování, problémy se soustředěním, neklid, změny školního prospěchu a hyperaktivitu.
3. NEURONTIN může způsobit závažné nebo život ohrožující alergické reakce které mohou ovlivnit vaši kůži nebo jiné části vašeho těla, jako jsou vaše játra nebo krevní buňky. To může způsobit, že budete hospitalizováni nebo vysazeni NEURONTIN. Můžete nebo nemusíte mít vyrážku s alergickou reakcí způsobenou přípravkem NEURONTIN. Okamžitě zavolejte poskytovatele zdravotní péče, pokud máte některý z následujících příznaků:
- vyrážka
- kopřivka
- potíže s dýcháním
- horečka
- oteklé žlázy, které nezmizí
- otok obličeje, rtů, hrdla nebo jazyka
- zežloutnutí kůže nebo očního bělma
- neobvyklé modřiny nebo krvácení
- silná únava nebo slabost
- neočekávaná bolest svalů
- časté infekce
Tyto příznaky mohou být prvními příznaky závažné reakce. Poskytovatel zdravotní péče by vás měl vyšetřit, aby rozhodl, zda máte pokračovat v užívání přípravku NEURONTIN.
4. Vážné problémy s dýcháním. Závažné problémy s dýcháním se mohou objevit, pokud je NEURONTIN užíván s jinými léky, které mohou způsobit těžkou ospalost nebo sníženou pozornost, nebo když jej užívá někdo, kdo již má problémy s dýcháním. Při zahájení léčby přípravkem NEURONTIN 300 mg nebo při zvýšení dávky sledujte zvýšenou ospalost nebo snížené dýchání. Pokud se vyskytnou dýchací potíže, okamžitě vyhledejte pomoc.
Co je NEURONTIN 400 mg?
NEURONTIN 400 mg je lék na předpis používaný k léčbě:
- Bolest z poškozených nervů (postherpetická bolest), která následuje po hojení pásového oparu (bolestivá vyrážka, která přichází po infekci herpes zoster) u dospělých.
- Částečné záchvaty při současném užívání s jinými léky u dospělých a dětí ve věku 3 let a starších se záchvaty.
Kdo by neměl užívat NEURONTIN?
Neužívejte přípravek NEURONTIN, jestliže jste alergický(á) na gabapentin nebo na kteroukoli další složku přípravku NEURONTIN. Úplný seznam složek přípravku NEURONTIN naleznete na konci tohoto Průvodce léčivy.
Co mám sdělit svému poskytovateli zdravotní péče před užitím přípravku NEURONTIN 300 mg?
Před užitím přípravku NEURONTIN 400 mg informujte svého poskytovatele zdravotní péče, pokud:
- máte nebo jste měl(a) problémy s ledvinami nebo jste na hemodialýze
- máte nebo jste měli deprese, problémy s náladou nebo sebevražedné myšlenky nebo chování
- mít cukrovku
- mít problémy s dýcháním
- jste těhotná nebo plánujete otěhotnět. Není známo, zda NEURONTIN může poškodit vaše nenarozené dítě. Okamžitě informujte svého poskytovatele zdravotní péče, pokud otěhotníte během užívání přípravku NEURONTIN. Vy a váš poskytovatel zdravotní péče se rozhodnete, zda máte užívat přípravek NEURONTIN 100 mg, když jste těhotná. Registr těhotenství: Pokud otěhotníte během užívání přípravku NEURONTIN, promluvte si se svým poskytovatelem zdravotní péče o registraci v Severoamerickém registru antiepileptických léků (NAAED) Pregnancy Registry. Účelem tohoto registru je shromažďovat informace o bezpečnosti antiepileptik během těhotenství. Do tohoto registru se můžete zapsat na telefonním čísle 1-888-233-2334.
- kojíte nebo plánujete kojit. NEURONTIN může přecházet do mateřského mléka. Vy a váš poskytovatel zdravotní péče byste se měli rozhodnout, jak budete krmit své dítě, když užíváte NEURONTIN.
Informujte svého poskytovatele zdravotní péče o všech lécích, které užíváte, včetně léků na předpis a volně prodejných léků, vitamínů a bylinných doplňků. Zejména informujte svého poskytovatele zdravotní péče, pokud užíváte jakýkoli opioidní lék proti bolesti (jako je oxykodon), jakékoli léky na úzkost (jako je lorazepam) nebo nespavost (jako je zolpidem) nebo jakékoli léky, které způsobují ospalost.
Pokud tyto léky užíváte s přípravkem NEURONTIN, můžete mít vyšší riziko závratí, ospalosti nebo dýchacích problémů.
Užívání přípravku NEURONTIN 100 mg s některými jinými léky může způsobit nežádoucí účinky nebo ovlivnit jejich účinnost. Nezačínejte ani nevysazujte jiné léky, aniž byste si promluvili se svým poskytovatelem zdravotní péče.
Znát léky, které užíváte. Uchovávejte si jejich seznam a ukažte jej svému poskytovateli zdravotní péče a lékárníkovi, když dostanete nový lék.
Jak mám užívat NEURONTIN 400 mg?
- Užívejte NEURONTIN přesně podle předpisu. Váš poskytovatel zdravotní péče vám sdělí, jaké množství přípravku NEURONTIN 600 mg máte užívat.
- Neměňte dávku přípravku NEURONTIN, aniž byste si promluvili se svým poskytovatelem zdravotní péče.
- Pokud užijete tablety NEURONTIN a rozlomíte tabletu na polovinu, nepoužitá polovina tablety by měla být užita v další plánované dávce. Půlky tablety nepoužité do 28 dnů po rozlomení je třeba zlikvidovat.
- Kapsle NEURONTIN zapijte vodou.
- Tablety NEURONTIN lze užívat s jídlem nebo bez jídla. Pokud užíváte antacidum obsahující hliník a hořčík, jako je Maalox®, Mylanta®, Gelusil®, Gaviscon® nebo Di-Gel®, měli byste počkat alespoň 2 hodiny, než si užijete další dávku přípravku NEURONTIN. Pokud užijete příliš mnoho přípravku NEURONTIN 600 mg, ihned zavolejte svého poskytovatele zdravotní péče nebo místní Poison Control Center na čísle 1-800-222-1222.
Čemu se mám vyvarovat při užívání přípravku NEURONTIN?
- Během užívání přípravku NEURONTIN nepijte alkohol ani neužívejte jiné léky, které způsobují ospalost nebo závratě, aniž byste si nejprve promluvili se svým poskytovatelem zdravotní péče. Užívání přípravku NEURONTIN 600 mg s alkoholem nebo léky, které způsobují ospalost nebo závratě, může vaši ospalost nebo závratě zhoršit.
- Neřiďte, neobsluhujte těžké stroje ani neprovádějte jiné nebezpečné činnosti, dokud nezjistíte, jak na vás NEURONTIN 100 mg působí. NEURONTIN 600 mg může zpomalit vaše myšlení a motoriku.
Jaké jsou možné nežádoucí účinky přípravku NEURONTIN 300 mg?
NEURONTIN 100 mg může způsobit závažné nežádoucí účinky včetně:
Vidět "Jaké jsou nejdůležitější informace, které bych měl vědět o NEURONTIN?"
- problémy s řízením při používání přípravku NEURONTIN. Viz "Čemu bych se měl vyvarovat při užívání Neurontinu 600 mg?"
- ospalost a závratě, které by mohly zvýšit výskyt náhodných zranění, včetně pádů
- Mezi nejčastější nežádoucí účinky přípravku NEURONTIN 100 mg patří:
- nedostatek koordinace
- virová infekce
- pocit ospalosti
- nevolnost a zvracení
- potíže s mluvením
- třes
- otoky, obvykle nohou a chodidel
- cítit se unaveně
- horečka
- trhavé pohyby
- potíže s koordinací
- dvojité vidění
- neobvyklý pohyb očí
Informujte svého poskytovatele zdravotní péče, pokud máte jakýkoli vedlejší účinek, který vás obtěžuje nebo který nezmizí.
To nejsou všechny možné vedlejší účinky přípravku NEURONTIN. Další informace získáte od svého poskytovatele zdravotní péče nebo lékárníka.
Zavolejte svého lékaře o radu ohledně nežádoucích účinků. Nežádoucí účinky můžete hlásit úřadu FDA na čísle 1-800-FDA-1088.
Jak mám uchovávat NEURONTIN?
- Uchovávejte NEURONTIN 400 mg tobolky a tablety při teplotě od 68 °F do 77 °F (20 °C až 25 °C).
- Uchovávejte NEURONTIN 400 mg perorální roztok v chladničce při teplotě mezi 36 °F až 46 °F (2 °C až 8 °C).
Uchovávejte NEURONTIN 600 mg a všechny léky mimo dosah dětí.
Obecné informace o bezpečném a účinném používání přípravku NEURONTIN
Léky jsou někdy předepisovány pro jiné účely, než které jsou uvedeny v Průvodci léky. Neužívejte přípravek NEURONTIN 300 mg na stav, pro který nebyl předepsán. Nedávejte NEURONTIN jiným lidem, i když mají stejné příznaky jako vy. Může jim to ublížit.
Tento průvodce léky shrnuje nejdůležitější informace o přípravku NEURONTIN. Pokud chcete další informace, promluvte si se svým poskytovatelem zdravotní péče. Můžete požádat svého poskytovatele zdravotní péče nebo lékárníka o informace o přípravku NEURONTIN 100 mg, který byl napsán pro zdravotnické pracovníky.
Pro více informací navštivte http://www.pfizer.com nebo volejte 1-800-438-1985.
Jaké jsou složky přípravku NEURONTIN 400 mg?
Aktivní složka: gabapentin
Neaktivní složky v kapslích: laktóza, kukuřičný škrob, mastek, želatina, oxid titaničitý a FD&C Blue č. 2.
Obal tobolky 300 mg také obsahuje: žlutý oxid železitý.
Obal tobolky 400 mg také obsahuje: červený oxid železitý a žlutý oxid železitý.
Neaktivní složky v tabletách: poloxamer 407, kopovidon, kukuřičný škrob, stearát hořečnatý, hydroxypropylcelulóza, mastek a kandelilový vosk
Neaktivní složky v perorálním roztoku: glycerin, xylitol, čištěná voda a umělé aroma.
Tato příručka pro léky byla schválena americkým Úřadem pro kontrolu potravin a léčiv.