Omnicef 300mg Cefdinir Použití, vedlejší účinky a dávkování. Cena v internetové lékárně. Generické léky bez předpisu.
Co je Omnicef a jak se používá?
Omnicef (cefdinir) je cefalosporinové antibiotikum používané k léčbě mnoha různých typů infekcí způsobených bakteriemi. Značka Omnicef je v USA ukončena Omnicef je k dispozici v obecné formě.
Jaké jsou vedlejší účinky Omnicefu?
Mezi časté nežádoucí účinky přípravku Omnicef 300 mg patří:
- průjem,
- nevolnost,
- zvracení,
- bolest břicha,
- špatné trávení,
- bolest hlavy,
- závrať,
- plenková vyrážka u kojence užívajícího tekutý cefdinir,
- svědění, popř
- vyrážka,
Poznámka: Tento seznam nemusí obsahovat všechny možné vedlejší účinky.
Aby se omezil vývoj bakterií rezistentních vůči lékům a zachovala se účinnost přípravku OMNICEF 300 mg a dalších antibakteriálních léků, měl by být přípravek OMNICEF používán pouze k léčbě nebo prevenci infekcí, u kterých je prokázáno nebo existuje silné podezření, že jsou způsobeny bakteriemi.
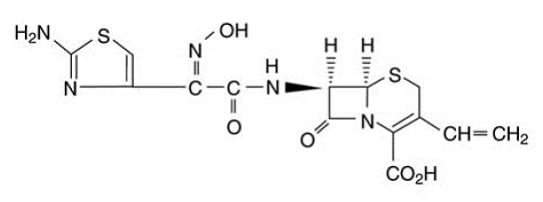
POPIS
Kapsle OMNICEF (cefdinir) a OMNICEF (cefdinir) pro perorální suspenzi obsahují účinnou látku cefdinir, semisyntetický cefalosporin s rozšířeným spektrem pro perorální podání. Chemicky je cefdinir [6R-[6a,7p(Z)]-7-[[(2-amino-4-thiazolyl)(hydroxyimino)acetyl]amino]3-ethenyl-8-oxo-5-thia-1 -azabicyklo[4.2.0]okt-2-en-2-karboxylová kyselina. Cefdinir je bílá až slabě hnědožlutá pevná látka. Je mírně rozpustný ve zředěné kyselině chlorovodíkové a mírně rozpustný v 0,1 M fosfátovém pufru o pH 7,0. Empirický vzorec je C14H13N5O5S2 a molekulová hmotnost je 395,42. Cefdinir má strukturní vzorec uvedený níže:
OMNICEF 300 mg tobolky obsahují 300 mg cefdinir a následující neaktivní složky: karboxymethylcelulóza vápenatá, NF; polyoxyl 40 stearát, NF; a stearát hořečnatý, NF. Obaly tobolek obsahují FD&C Blue #1; FD&C Red #40; D&C Red #28; oxid titaničitý, NF; želatina, NF; oxid křemičitý, NF; a laurylsulfát sodný, NF.
OMNICEF 300 mg pro perorální suspenzi po rekonstituci obsahuje 125 mg cefdiniru na 5 ml nebo 250 mg cefdiniru na 5 ml a následující neaktivní složky: sacharózu, NF; kyselina citrónová, USP; citrát sodný, USP; benzoát sodný, NF; xantanová guma, NF; guarová guma, NF; umělé jahodové a smetanové příchutě; oxid křemičitý, NF; a stearát hořečnatý, NF.
INDIKACE
Aby se snížil vývoj bakterií rezistentních vůči lékům a zachovala se účinnost přípravku OMNICEF 300 mg a dalších antibakteriálních léků, měl by být přípravek OMNICEF 300 mg používán pouze k léčbě nebo prevenci infekcí, u kterých je prokázáno nebo existuje silné podezření, že jsou způsobeny citlivými bakteriemi. Pokud jsou k dispozici informace o kultivaci a citlivosti, je třeba je vzít v úvahu při výběru nebo úpravě antibakteriální terapie. Při absenci takových údajů mohou k empirickému výběru terapie přispět místní epidemiologie a vzorce citlivosti.
OMNICEF (cefdinir) tobolky a OMNICEF (cefdinir) pro perorální suspenzi jsou indikovány k léčbě pacientů s mírnými až středně těžkými infekcemi způsobenými citlivými kmeny určených mikroorganismů za níže uvedených stavů.
Dospělí a dospívající
Pneumonie získaná v komunitě
způsobené Haemophilus influenzae (včetně kmenů produkujících β-laktamázu), Haemophilus parainfluenzae (včetně kmenů produkujících β-laktamázu), Streptococcus pneumoniae (pouze kmeny citlivé na penicilin) a Moraxella catarrhalis (včetně kmenů produkujících β-laktamázu) (viz. Klinické studie ).
Akutní exacerbace chronické bronchitidy
způsobené Haemophilus influenzae (včetně kmenů produkujících β-laktamázu), Haemophilus parainfluenzae (včetně kmenů produkujících β-laktamázu), Streptococcus pneumoniae (pouze kmeny citlivé na penicilin) a Moraxella catarrhalis (včetně kmenů produkujících β-laktamázu).
Akutní maxilární sinusitida
způsobené Haemophilus influenzae (včetně kmenů produkujících β-laktamázu), Streptococcus pneumoniae (pouze kmeny citlivé na penicilin) a Moraxella catarrhalis (včetně kmenů produkujících β-laktamázu).
POZNÁMKA: Informace o použití u dětských pacientů viz Pediatrické použití a DÁVKOVÁNÍ A PODÁVÁNÍ .
Faryngitida/tonzilitida
způsobené Streptococcus pyogenes (viz Klinické studie ).
POZNÁMKA: Cefdinir je účinný při eradikaci S. pyogenes z orofaryngu. Cefdinir však nebyl studován pro prevenci revmatické horečky po faryngitidě/tonzilitidě způsobené S. pyogenes. Pouze intramuskulární penicilin byl prokázán jako účinný pro prevenci revmatické horečky.
Nekomplikované infekce kůže a kožní struktury
způsobené Staphylococcus aureus (včetně kmenů produkujících β-laktamázu) a Streptococcus pyogenes.
Pediatričtí pacienti
Akutní bakteriální zánět středního ucha způsobené Haemophilus influenzae (včetně kmenů produkujících β-laktamázu), Streptococcus pneumoniae (pouze kmeny citlivé na penicilin) a Moraxella catarrhalis (včetně kmenů produkujících β-laktamázu).
Faryngitida/tonzilitida
způsobené Streptococcus pyogenes (viz Klinické studie ).
POZNÁMKA: Cefdinir je účinný při eradikaci S. pyogenes z orofaryngu. Cefdinir však nebyl studován pro prevenci revmatické horečky po faryngitidě/tonzilitidě způsobené S. pyogenes. Pouze intramuskulární penicilin byl prokázán jako účinný pro prevenci revmatické horečky.
Nekomplikované infekce kůže a kožní struktury
způsobené Staphylococcus aureus (včetně kmenů produkujících β-laktamázu) a Streptococcus pyogenes.
DÁVKOVÁNÍ A PODÁVÁNÍ
(vidět INDIKACE A POUŽITÍ pro Indikované patogeny )
Kapsle
Doporučené dávkování a délka léčby infekcí u dospělých a dospívajících jsou popsány v následující tabulce; celková denní dávka pro všechny infekce je 600 mg. Dávkování jednou denně po dobu 10 dnů je stejně účinné jako dávkování BID. Dávkování jednou denně nebylo studováno u pneumonie nebo kožních infekcí; proto by měly být OMNICEF tobolky u těchto infekcí podávány dvakrát denně. Kapsle OMNICEF lze užívat bez ohledu na jídlo.
Dospělí a dospívající (věk 13 let a starší)
Prášek pro perorální suspenzi
Doporučené dávkování a délka léčby infekcí u dětských pacientů jsou popsány v následující tabulce; celková denní dávka pro všechny infekce je 14 mg/kg až do maximální dávky 600 mg denně. Dávkování jednou denně po dobu 10 dnů je stejně účinné jako dávkování BID. Dávkování jednou denně nebylo studováno u kožních infekcí; proto by měl být OMNICEF 300 mg pro perorální suspenzi u této infekce podáván dvakrát denně. OMNICEF pro perorální suspenzi lze podávat bez ohledu na jídlo.
Pediatričtí pacienti (věk 6 měsíců až 12 let)
OMNICEF PRO PERORÁLNÍ SUSPENZI PEDIATRICKÝ DÁVKOVÁNÍ TABULKA
Pacienti s renální insuficiencí
U dospělých pacientů s clearance kreatininu
ambulantních pacientů je obtížné měřit clearance kreatininu. K odhadu clearance kreatininu (CLcr) u dospělých pacientů však lze použít následující vzorec. Aby byly odhady platné, hladiny kreatininu v séru by měly odrážet ustálené hladiny renálních funkcí.
kde clearance kreatininu je v ml/min, věk je v letech, hmotnost je v kilogramech a sérový kreatinin je v mg/dl.4
K odhadu clearance kreatininu u pediatrických pacientů lze použít následující vzorec:
CLcr = K × délka těla nebo výška/sérový kreatinin
kde K = 0,55 pro dětské pacienty starší 1 roku5 a 0,45 pro kojence (do 1 roku)6.
Ve výše uvedené rovnici je clearance kreatininu v ml/min/1,73 m², délka těla nebo výška je v centimetrech a sérový kreatinin je v mg/dl.
U pediatrických pacientů s clearance kreatininu
Pacienti na hemodialýze
Hemodialýza odstraňuje cefdinir z těla. U pacientů udržovaných na chronické hemodialýze je doporučený počáteční dávkovací režim 300 mg nebo 7 mg/kg každý druhý den.
Na závěr každé hemodialýzy by se mělo podat 300 mg (nebo 7 mg/kg). Následné dávky (300 mg nebo 7 mg/kg) se pak podávají každý druhý den.
Pokyny pro míchání Omnicef 300 mg pro perorální suspenzi
Po smíchání může být suspenze skladována při pokojové teplotě (25°C/77°F). Nádoba by měla být těsně uzavřena a suspenze by měla být před každým podáním dobře protřepána. Suspenze může být používána po dobu 10 dnů, poté musí být jakákoli nepoužitá část zlikvidována.
JAK DODÁVÁNO
Kapsle OMNICEF , obsahující 300 mg cefdiniru, jako levandulové a tyrkysové tobolky s potiskem názvu přípravku, jsou dostupné takto:
60 kapslí/láhev NDC 0074-3769-60 OMNI-PAC™ karton se 3 blistrovými kartami na 5 dní a 10 tobolkami pro použití NDC 0074-3769-30
OMNICEF 300 mg pro perorální suspenzi je krémově zbarvený prášek, který po rekonstituci podle návodu obsahuje 125 mg cefdinir/5 ml nebo 250 mg cefdinir/5 ml. Rekonstituované suspenze mají krémovou barvu a jahodovou příchuť. Prášek je dostupný následovně:
125 mg/5 ml
60ml lahvičky NDC 0074-3771-60 100ml lahvičky NDC 0074-3771-13 250 mg/5 ml 60ml lahvičky NDC 0074-6151-60 100ml lahvičky NDC 0074-6151-13
Uchovávejte tobolky a nesuspendovaný prášek při teplotě 25 °C (77 °F); povolené odchylky do 15°-30°C (59°-86°F) [viz USP Controlled Room Temperature]. Po rekonstituci lze perorální suspenzi uchovávat při kontrolované pokojové teplotě po dobu 10 dnů.
Výrobce: CEPH International Corporation Carolina, Puerto Rico 00986. Pro: AbbVie Inc., North Chicago, IL 60064, USA, v licenci: Astellas Pharma Inc. Tokyo, Japonsko. Revize listopadu 2015
VEDLEJŠÍ EFEKTY
Nežádoucí příhody
Klinické studie – OMNICEF tobolky (dospělí a dospívající pacienti)
V klinických studiích bylo 5 093 dospělých a dospívajících pacientů (3 841 v USA a 1 252 mimo USA) léčeno doporučenou dávkou tobolek cefdiniru (600 mg/den). Většina nežádoucích účinků byla mírná a sama odezněla. Cefdiniru nebyla připisována žádná úmrtí ani trvalé postižení. Sto čtyřicet sedm z 5 093 (3 %) pacientů přerušilo medikaci kvůli nežádoucím účinkům, o nichž se výzkumníci domnívali, že jsou možná, pravděpodobně nebo určitě spojeny s terapií cefdinirem. Přerušení byla primárně pro gastrointestinální poruchy, obvykle průjem nebo nevolnost. Devatenáct z 5093 (0,4 %) pacientů bylo přerušeno z důvodu vyrážky související s podáváním cefdiniru.
Ve Spojených státech se následující nežádoucí příhody v klinických studiích s opakovanými dávkami (N = 3 841 pacientů léčených cefdinirem) výzkumníci domnívali, že možná, pravděpodobně nebo určitě souvisejí s tobolkami cefdiniru:
NEŽÁDOUCÍ UDÁLOSTI SPOJENÉ S KAPSLEMI CEFDINIR ZKOUŠKY U DOSPĚLÝCH A DOSPĚLÝCH PACIENTŮ (N = 3841)a
Během klinických studií provedených v USA byly pozorovány následující změny laboratorních hodnot s možným klinickým významem, bez ohledu na vztah k léčbě cefdinirem:
ZMĚNY HODNOTY LABORATOŘE POZOROVANÉ U TABLES CEFDINIR ZKOUŠKY U DOSPĚLÝCH A DOSPĚLÝCH PACIENTŮ (N = 3841)
Klinické zkoušky – OMNICEF pro perorální suspenzi (dětští pacienti)
klinických studiích bylo doporučenou dávkou suspenze cefdiniru (14 mg/kg/den) léčeno 2289 pediatrických pacientů (1783 pacientů z USA a 506 pacientů mimo USA). Většina nežádoucích účinků byla mírná a sama odezněla. Cefdiniru nebyla připisována žádná úmrtí ani trvalé postižení. Čtyřicet z 2 289 (2 %) pacientů přerušilo medikaci kvůli nežádoucím účinkům, které výzkumníci považovali za možná, pravděpodobně nebo určitě související s léčbou cefdinirem. Přerušení byla primárně pro gastrointestinální poruchy, obvykle průjem. Pět z 2289 (0,2 %) pacientů bylo přerušeno z důvodu vyrážky související s podáváním cefdiniru.
V USA se výzkumníci domnívali, že následující nežádoucí příhody jsou možná, pravděpodobně nebo určitě související se suspenzí cefdiniru v klinických studiích s opakovanými dávkami (N = 1783 pacientů léčených cefdinirem):
NEŽÁDOUCÍ UDÁLOSTI SPOJENÉ S POZASTAVENÍM CEFDINIRU US TESTY U PEDIATRICKÝCH PACIENTŮ (N = 1783)a
POZNÁMKA: U pacientů léčených cefdinirem i kontrolní skupinou byl výskyt průjmu a vyrážky vyšší u nejmladších dětských pacientů. Incidence průjmu u pacientů ve věku ≤ 2 roky léčených cefdinirem byla 17 % (95/557) ve srovnání se 4 % (51/1226) u pacientů starších 2 let. Výskyt vyrážky (především plenkové vyrážky u mladších pacientů) byl 8 % (43/557) u pacientů ve věku ≤ 2 roky ve srovnání s 1 % (8/1226) u pacientů starších 2 let.
Během klinických studií provedených v USA byly pozorovány následující změny laboratorních hodnot s možným klinickým významem, bez ohledu na vztah k léčbě cefdinirem:
ZMĚNY LABORATORNÍ HODNOTY MOŽNÉHO KLINICKÉHO VÝZNAMU POZOROVANÉ PŘI ZKOUŠKÁCH USPOUŠTĚNÍ CEFDINIRU U PEDIATRICKÝCH PACIENTŮ (N = 1783)
Postmarketingové zkušenosti
Následující nežádoucí účinky a změněné laboratorní testy, bez ohledu na jejich vztah k cefdiniru, byly hlášeny během rozsáhlých postmarketingových zkušeností, počínaje schválením v Japonsku v roce 1991: šok, anafylaxe se vzácnými případy úmrtí, edém obličeje a hrtanu, pocit dušení, reakce podobné sérové nemoci, konjunktivitida, stomatitida, Stevens-Johnsonův syndrom, toxická epidermální nekrolýza, exfoliativní dermatitida, erythema multiforme, erythema nodosum, akutní hepatitida, cholestáza, fulminantní hepatitida, selhání jater, žloutenka, zvýšená amyláza, akutní enteroitida, hemoragická kolitida, meléna, pseudomembranózní kolitida, pancytopenie, granulocytopenie, leukopenie, trombocytopenie, idiopatická trombocytopenická purpura, hemolytická anémie, akutní respirační selhání, astmatický záchvat, pneumonie vyvolaná léky, eozinofilní pneumonie, idiopatická intersticiální pneumonie, horečka, akutní renální selhání sklon ke krvácení, koagulační dis řádu, diseminovaná intravaskulární koagulace, krvácení do horní části GI, peptický vřed, ileus, ztráta vědomí, alergická vaskulitida, možná interakce cefdinir-diclofenac, srdeční selhání, bolest na hrudi, infarkt myokardu, hypertenze, mimovolní pohyby a rhabdomyolýza.
Nežádoucí účinky třídy cefalosporinů
U antibiotik třídy cefalosporinů byly obecně hlášeny následující nežádoucí účinky a změněné laboratorní testy:
Alergické reakce, anafylaxe, Stevens-Johnsonův syndrom, erythema multiforme, toxická epidermální nekrolýza, renální dysfunkce, toxická nefropatie, jaterní dysfunkce včetně cholestázy, aplastická anémie, hemolytická anémie, krvácení, falešně pozitivní test na glukózu v moči, neutropenie a pancytopenie, . Příznaky pseudomembranózní kolitidy se mohou objevit během nebo po léčbě antibiotiky (viz VAROVÁNÍ ).
Některé cefalosporiny se podílejí na spouštění záchvatů, zejména u pacientů s poruchou funkce ledvin, kdy dávka nebyla snížena (viz DÁVKOVÁNÍ A PODÁVÁNÍ a PŘEDÁVKOVÁNÍ ). Pokud se objeví záchvaty spojené s lékovou terapií, lék by měl být vysazen. Pokud je to klinicky indikováno, lze podat antikonvulzivní léčbu.
DROGOVÉ INTERAKCE
Antacida (obsahující hliník nebo hořčík)
Současné podávání 300mg tobolek cefdiniru s 30 ml suspenze Maalox® TC snižuje rychlost (Cmax) a rozsah (AUC) absorpce přibližně o 40 %. Doba do dosažení Cmax se také prodlouží o 1 hodinu. Pokud je antacidum podáno 2 hodiny před nebo 2 hodiny po cefdiniru, nedochází k žádným významným účinkům na farmakokinetiku cefdiniru. Pokud jsou během léčby přípravkem OMNICEF 300 mg potřeba antacida, měl by se přípravek OMNICEF 300 mg užívat alespoň 2 hodiny před nebo po podání antacida.
Probenecid
Stejně jako u jiných β-laktamových antibiotik i probenecid inhibuje renální vylučování cefdiniru, což má za následek přibližné zdvojnásobení AUC, 54% zvýšení maximálních plazmatických hladin cefdiniru a 50% prodloužení zdánlivého eliminačního t½.
Doplňky železa a potraviny obohacené železem
Současné podávání cefdiniru s terapeutickým doplňkem železa obsahujícím 60 mg elementárního železa (jako FeSO4) nebo vitaminy doplněné 10 mg elementárního železa snížilo rozsah absorpce o 80 % a 31 %. Pokud jsou během léčby přípravkem OMNICEF 300 mg vyžadovány doplňky železa, měl by se přípravek OMNICEF užívat alespoň 2 hodiny před nebo po podání.
Vliv potravin vysoce obohacených elementárním železem (především snídaňové cereálie obohacené železem) na absorpci cefdiniru nebyl studován.
Současné podávání kojenecké výživy obohacené železem (2,2 mg elementárního železa/6 uncí) nemá žádný významný vliv na farmakokinetiku cefdiniru. Proto lze OMNICEF pro perorální suspenzi podávat s kojeneckou výživou obohacenou železem.
pacientů užívajících cefdinir byly hlášeny případy načervenalé stolice. V mnoha případech pacienti dostávali také produkty obsahující železo. Načervenalá barva je způsobena tvorbou nevstřebatelného komplexu mezi cefdinirem nebo jeho produkty rozkladu a železem v gastrointestinálním traktu.
Interakce drog/laboratorních testů
Falešně pozitivní reakce na ketony v moči se může objevit u testů s použitím nitroprusidu, ale ne u testů používajících nitroferricyanid. Podávání cefdiniru může mít za následek falešně pozitivní reakci na glukózu v moči při použití Clinitest®, Benedictova roztoku nebo Fehlingova roztoku. Doporučuje se používat glukózové testy založené na enzymatických glukózooxidázových reakcích (jako je Clinistix® nebo Tes-Tape®). Je známo, že cefalosporiny příležitostně vyvolávají pozitivní přímý Coombsův test.
VAROVÁNÍ
PŘED ZAHÁJENÍM TERAPIE OMNICEFEM (CEFDINIR) BY MĚLO BÝT PROVEDENO PEČLIVÉHO ŠETŘENÍ A ZJIŠTĚNO, ZDA PACIENT MĚL PŘEDCHOZÍ REAKCE PŘECENITELNOSTI NA CEFDINIR, JINÉ CEPHALOSPORINY, GSOR.JINÉ PENICILINY POKUD JE CEFDINIR PODÁVÁN PACIENTŮM CITLIVÝM NA PENICILIN, JE NUTNÉ ZVYŠOVAT OPATRNOST, PROTOŽE ZKRÍŽOVÁ HYPERENZITIVITA MEZI β-LAKTAMOVÝMI ANTIBIOTIKY JSOU JASNĚ DOKUMENTOVÁNY A MŮŽE SE VYSKYTnout AŽ 10 % VZNIKU PENYA AŽ VE VZNIKU PWI. POKUD SE DOSTANE ALERGICKÁ REAKCE NA CEFDINIR, JE TŘEBA LÉK UKONČIT. VÁŽNÉ AKUTNÍ REAKCE PŘECITLIVOSTI MŮŽE VYŽADOVAT LÉČBA EPINEPRINEM A JINÁ NOUZOVÁ OPATŘENÍ, VČETNĚ KYSLÍKU, INTRAVENÓZNÍCH TEKUTIN, INTRAVENOZNÍCH ANTIHISTAMINIEK, KORTIKOSTEROIDŮ, AMINŮ PRESSORŮ A SYSTÉMU ŘÍZENÍ VZDUCHU CLINICAL.
Průjem spojený s Clostridium difficile (CDAD) byl hlášen při použití téměř všech antibakteriálních látek, včetně OMNICEF, a jeho závažnost se může pohybovat od mírného průjmu až po fatální kolitidu. Léčba antibakteriálními látkami mění normální flóru tlustého střeva, což vede k přemnožení C. difficile.
C. difficile produkuje toxiny A a B, které přispívají k rozvoji CDAD. Kmeny C. difficile produkující hypertoxin způsobují zvýšenou morbiditu a mortalitu, protože tyto infekce mohou být odolné vůči antimikrobiální léčbě a mohou vyžadovat kolektomii. CDAD je třeba vzít v úvahu u všech pacientů, kteří mají průjem po použití antibiotik. Je nutná pečlivá anamnéza, protože CDAD se objevilo více než dva měsíce po podání antibakteriálních látek.
Je-li podezření na CDAD nebo je-li potvrzeno, může být nutné přerušit pokračující používání antibakteriálních látek, které nejsou zaměřeny proti C. difficile. Podle klinické indikace by měla být zahájena vhodná léčba tekutin a elektrolytů, suplementace proteinů, antibakteriální léčba C. difficile a chirurgické vyšetření.
OPATŘENÍ
Všeobecné
Je nepravděpodobné, že by předepsání přípravku OMNICEF 300 mg v nepřítomnosti prokázané nebo silně suspektní bakteriální infekce nebo profylaktické indikace přineslo pacientovi prospěch a zvýšilo riziko rozvoje bakterií rezistentních na léky.
Stejně jako u jiných širokospektrých antibiotik může dlouhodobá léčba vést k možnému vzniku a přemnožení rezistentních organismů. Pečlivé sledování pacienta je nezbytné. Pokud se během léčby objeví superinfekce, měla by být podána vhodná alternativní léčba.
Cefdinir, stejně jako ostatní širokospektrá antimikrobiální látky (antibiotika), by měl být předepisován s opatrností u jedinců s kolitidou v anamnéze.
pacientů s přechodnou nebo přetrvávající renální insuficiencí (clearance kreatininu DÁVKOVÁNÍ A PODÁVÁNÍ ).
Karcinogeneze, mutageneze, zhoršení plodnosti
Karcinogenní potenciál cefdiniru nebyl hodnocen. Žádné mutagenní účinky nebyly pozorovány v testu bakteriální reverzní mutace (Ames) nebo testu bodových mutací na lokusu hypoxanthin-guanin fosforibosyltransferázy (HGPRT) v plicních buňkách čínského křečka V79. Žádné klastogenní účinky nebyly pozorovány in vitro v testu strukturních chromozomových aberací v plicních buňkách čínského křečka V79 ani in vivo v mikronukleovém testu v kostní dřeni myší. U potkanů nebyla fertilita a reprodukční schopnost ovlivněna cefdinirem v perorálních dávkách až do 1000 mg/kg/den (70násobek dávky pro člověka na mg/kg/den, 11násobek na mg/m2/den).
Těhotenství
Teratogenní účinky
Těhotenství kategorie B
Cefdinir nebyl teratogenní u potkanů při perorálních dávkách do 1000 mg/kg/den (70násobek dávky pro člověka na základě mg/kg/den, 11násobek na základě mg/m²/den) nebo u králíků při perorálních dávkách do 10 mg/kg/den (0,7násobek lidské dávky na mg/kg/den, 0,23násobek na mg/m2/den). Mateřská toxicita (snížený přírůstek tělesné hmotnosti) byla pozorována u králíků při maximální tolerované dávce 10 mg/kg/den bez nežádoucích účinků na potomstvo. Snížení tělesné hmotnosti se objevilo u plodů potkanů při dávce ≥ 100 mg/kg/den au potomků potkanů při dávce ≥ 32 mg/kg/den. Nebyly pozorovány žádné účinky na reprodukční parametry matek nebo přežití, vývoj, chování nebo reprodukční funkce potomků.
Neexistují však žádné adekvátní a dobře kontrolované studie u těhotných žen. Vzhledem k tomu, že reprodukční studie na zvířatech nejsou vždy prediktivní pro lidskou odpověď, měl by být tento lék používán během těhotenství pouze tehdy, je-li to nezbytně nutné.
Práce a dodání
Cefdinir nebyl studován pro použití během porodu a porodu.
Kojící matky
Po podání jednotlivých dávek 600 mg nebyl cefdinir detekován v lidském mateřském mléce.
Pediatrické použití
Bezpečnost a účinnost u novorozenců a kojenců mladších 6 měsíců nebyla stanovena. Použití cefdiniru k léčbě akutní maxilární sinusitidy u pediatrických pacientů (ve věku 6 měsíců až 12 let) je podpořeno důkazy z adekvátních a dobře kontrolovaných studií u dospělých a dospívajících, podobnou patofyziologií akutní sinusitidy u dospělých a dětských pacientů a srovnávací farmakokinetické údaje u pediatrické populace.
Geriatrické použití
Účinnost je srovnatelná u geriatrických pacientů a mladších dospělých. Zatímco cefdinir byl dobře snášen ve všech věkových skupinách, v klinických studiích se u geriatrických pacientů vyskytl nižší výskyt nežádoucích účinků, včetně průjmu, než u mladších dospělých. Úprava dávky u starších pacientů není nutná, pokud není výrazně snížena funkce ledvin (viz DÁVKOVÁNÍ A PODÁVÁNÍ ).
PŘEDÁVKOVAT
Informace o předávkování cefdinirem u lidí nejsou k dispozici. Ve studiích akutní toxicity na hlodavcích nevyvolala jediná perorální dávka 5600 mg/kg žádné nežádoucí účinky. Toxické známky a příznaky po předávkování jinými β-laktamovými antibiotiky zahrnovaly nauzeu, zvracení, epigastrické potíže, průjem a křeče. Hemodialýza odstraňuje cefdinir z těla. To může být užitečné v případě závažné toxické reakce z předávkování, zejména pokud je snížena funkce ledvin.
KONTRAINDIKACE
OMNICEF (cefdinir) je kontraindikován u pacientů se známou alergií na cefalosporinovou třídu antibiotik.
KLINICKÁ FARMAKOLOGIE
Farmakokinetika a metabolismus léčiv
Vstřebávání
Orální biologická dostupnost
Maximální plazmatické koncentrace cefdiniru se objevují 2 až 4 hodiny po podání tobolky nebo suspenze. Plazmatické koncentrace cefdiniru se zvyšují s dávkou, ale zvýšení jsou méně než úměrná dávce od 300 mg (7 mg/kg) do 600 mg (14 mg/kg). Po podání suspenze zdravým dospělým je biologická dostupnost cefdiniru 120 % ve srovnání s tobolkami. Odhadovaná biologická dostupnost tobolek cefdiniru je 21 % po podání dávky 300 mg tobolky a 16 % po podání dávky 600 mg tobolky. Odhadovaná absolutní biologická dostupnost suspenze cefdiniru je 25 %. Bylo prokázáno, že perorální suspenze Cefdinir o síle 250 mg/5 ml je bioekvivalentní síle 125 mg/5 ml u zdravých dospělých nalačno.
Účinek jídla
Cmax a AUC cefdiniru z tobolek jsou sníženy o 16 % a 10 %, pokud jsou podávány s jídlem s vysokým obsahem tuků. U dospělých, kterým byla podávána perorální suspenze 250 mg/5 ml s jídlem s vysokým obsahem tuku, se Cmax a AUC cefdiniru snížily o 44 % a 33 %. Velikost těchto snížení pravděpodobně nebude klinicky významná, protože studie bezpečnosti a účinnosti perorální suspenze u pediatrických pacientů byly provedeny bez ohledu na příjem potravy. Proto lze cefdinir užívat bez ohledu na jídlo.
Cefdinir kapsle
Plazmatické koncentrace cefdiniru a hodnoty farmakokinetických parametrů po podání jedné perorální dávky 300 mg a 600 mg cefdiniru dospělým subjektům jsou uvedeny v následující tabulce:
Průměrné (± SD) hodnoty farmakokinetických parametrů cefdiniru v plazmě po podání tobolek dospělým subjektům
Suspenze Cefdinir
Plazmatické koncentrace cefdiniru a hodnoty farmakokinetických parametrů po podání jednorázové perorální dávky cefdiniru 7- a 14 mg/kg pediatrickým subjektům (ve věku 6 měsíců až 12 let) jsou uvedeny v následující tabulce:
Průměrné (± SD) hodnoty farmakokinetických parametrů cefdiniru v plazmě po podání suspenze pediatrickým subjektům
Vícenásobné dávkování
Cefdinir se po podávání jednou nebo dvakrát denně subjektům s normální funkcí ledvin neakumuluje v plazmě.
Rozdělení
Průměrný distribuční objem (Vdarea) cefdiniru u dospělých subjektů je 0,35 l/kg (± 0,29); u pediatrických subjektů (ve věku 6 měsíců až 12 let) je cefdinir Vdarea 0,67 l/kg (± 0,38). Cefdinir se ze 60 % až 70 % váže na plazmatické proteiny u dospělých i pediatrických subjektů; vazba je nezávislá na koncentraci.
Kožní puchýř
dospělých subjektů byl medián (rozmezí) maximálních koncentrací cefdiniru v blistrové tekutině 0,65 (0,33-1,1) a 1,1 (0,49-1,9) μg/ml pozorován 4 až 5 hodin po podání dávek 300 a 600 mg, v daném pořadí. Průměrné (± SD) hodnoty Cmax a AUC (0-∞) blistru byly 48 % (± 13) a 91 % (± 18) odpovídajících hodnot v plazmě.
Tkáň mandlí
U dospělých pacientů podstupujících elektivní tonzilektomii byly příslušné střední koncentrace cefdiniru v tkáni mandlí 4 hodiny po podání jednotlivých dávek 300 a 600 mg 0,25 (0,220,46) a 0,36 (0,22-0,80) μg/g. Průměrné koncentrace v tonzilové tkáni byly 24 % (± 8) odpovídajících plazmatických koncentrací.
Sinusová tkáň
dospělých pacientů podstupujících elektivní operaci maxilárního a etmoidního sinu byly příslušné střední koncentrace cefdiniru v tkáni cefdiniru 4 hodiny po podání jednotlivé dávky 300 a 600 mg
Plicní tkáň
U dospělých pacientů podstupujících diagnostickou bronchoskopii byly příslušné střední koncentrace cefdiniru v bronchiální sliznici 4 hodiny po podání jednotlivých dávek 300 a 600 mg 0,78 (
Tekutina středního ucha
14 pediatrických pacientů s akutním bakteriálním zánětem středního ucha byly příslušné střední koncentrace cefdiniru ve středoušní tekutině 3 hodiny po podání jednotlivých dávek 7 a 14 mg/kg 0,21 (
CSF
Údaje o pronikání cefdiniru do lidského mozkomíšního moku nejsou k dispozici.
Metabolismus a vylučování
Cefdinir není znatelně metabolizován. Aktivita je primárně způsobena mateřským lékem. Cefdinir je vylučován především ledvinami s průměrným poločasem eliminace z plazmy (t½) 1,7 (± 0,6) hodin. U zdravých jedinců s normální funkcí ledvin je renální clearance 2,0 (± 1,0) ml/min/kg a zdánlivá perorální clearance je 11,6 (± 6,0) a 15,5 (± 5,4) ml/min/kg po dávkách 300 a 600 -mg, resp. Průměrné procento dávky zjištěné v nezměněné podobě v moči po dávkách 300 mg a 600 mg je 18,4 % (± 6,4) a 11,6 % (± 4,6). Clearance cefdiniru je snížena u pacientů s renální dysfunkcí (viz Zvláštní populace - Pacienti s renální insuficiencí ).
Vzhledem k tomu, že renální exkrece je převládající cestou eliminace, mělo by být dávkování upraveno u pacientů s výrazně sníženou funkcí ledvin nebo u pacientů, kteří podstupují hemodialýzu (viz DÁVKOVÁNÍ A PODÁVÁNÍ ).
Zvláštní populace
Pacienti s renální insuficiencí
Farmakokinetika cefdiniru byla zkoumána u 21 dospělých subjektů s různým stupněm renálních funkcí. Snížení rychlosti eliminace cefdiniru, zdánlivé perorální clearance (CL/F) a renální clearance bylo přibližně úměrné snížení clearance kreatininu (CLcr). V důsledku toho byly plazmatické koncentrace cefdiniru vyšší a přetrvávaly déle u subjektů s poruchou funkce ledvin než u osob bez poruchy funkce ledvin. U subjektů s CLcr mezi 30 a 60 ml/min se Cmax a t½ zvýšily přibližně 2krát a AUC přibližně 3krát. U subjektů s CLcr DÁVKOVÁNÍ A PODÁVÁNÍ ).
Hemodialýza
Farmakokinetika cefdiniru byla studována u 8 dospělých subjektů podstupujících hemodialýzu. Dialýza (trvání 4 hodiny) odstranila 63 % cefdiniru z těla a snížila zdánlivý eliminační t½ z 16 (± 3,5) na 3,2 (± 1,2) hodin. U této populace pacientů se doporučuje úprava dávkování (viz DÁVKOVÁNÍ A PODÁVÁNÍ ).
Onemocnění jater
Vzhledem k tomu, že cefdinir je převážně eliminován ledvinami a není významně metabolizován, studie u pacientů s poruchou funkce jater nebyly provedeny. Nepředpokládá se, že u této populace bude nutná úprava dávkování.
Geriatričtí pacienti
Vliv věku na farmakokinetiku cefdiniru po jednorázové dávce 300 mg byl hodnocen u 32 subjektů ve věku 19 až 91 let. Systémová expozice cefdiniru byla podstatně zvýšena u starších subjektů (N = 16), Cmax o 44 % a AUC o 86 %. Toto zvýšení bylo způsobeno snížením clearance cefdiniru. Zdánlivý distribuční objem byl také snížen, takže nebyly pozorovány žádné znatelné změny zdánlivého eliminačního t½ (starší osoby: 2,2 ± 0,6 hodiny vs. mladé: 1,8 ± 0,4 hodiny). Protože se ukázalo, že clearance cefdiniru primárně souvisí spíše se změnami funkce ledvin než s věkem, starší pacienti nevyžadují úpravu dávkování, pokud nemají výrazně sníženou funkci ledvin (clearance kreatininu Pacienti s renální insuficiencí, výše ).
Pohlaví a rasa
Výsledky metaanalýzy klinické farmakokinetiky (N = 217) neukázaly žádný významný vliv pohlaví nebo rasy na farmakokinetiku cefdiniru.
Mikrobiologie
Mechanismus působení
Stejně jako u jiných cefalosporinů je baktericidní aktivita cefdiniru výsledkem inhibice syntézy buněčné stěny. Cefdinir je stabilní v přítomnosti některých, ale ne všech enzymů β-laktamázy. V důsledku toho je mnoho organismů odolných vůči penicilinům a některým cefalosporinům citlivých na cefdinir.
Mechanismus odporu
Rezistence na cefdinir je primárně způsobena hydrolýzou některými β-laktamázami, změnou proteinů vázajících penicilin (PBP) a sníženou permeabilitou. Cefdinir je neaktivní proti většině kmenů Enterobacter spp., Pseudomonas spp., Enterococcus spp., streptokokům rezistentním na penicilin a stafylokokům rezistentním na meticilin. β-laktamáza negativní, ampicilin-rezistentní (BLNAR) Kmeny H. influenzae jsou typicky necitlivé na cefdinir.
Antimikrobiální aktivita
Bylo prokázáno, že Cefdinir je účinný proti většině kmenů následujících mikroorganismů, a to jak in vitro, tak při klinických infekcích, jak je popsáno v INDIKACE A POUŽITÍ .
Gram-pozitivní bakterie
Staphylococcus aureus (pouze kmeny citlivé na meticilin) Streptococcus pneumoniae (pouze kmeny citlivé na penicilin) Streptococcus pyogenes
Gram-negativní bakterie
Haemophilus influenzae Haemophilus parainfluenzae Moraxella catarrhalis
K dispozici jsou následující údaje in vitro, ale jejich klinický význam není znám.
Cefdinir vykazuje in vitro minimální inhibiční koncentrace (MIC) 1 mcg/ml nebo méně proti (≥ 90 %) kmenům následujících mikroorganismů; bezpečnost a účinnost cefdiniru při léčbě klinických infekcí způsobených těmito mikroorganismy však nebyla stanovena v adekvátních a dobře kontrolovaných klinických studiích.
Gram-pozitivní bakterie
Staphylococcus epidermidis (pouze kmeny citlivé na meticilin) Streptococcus agalactiae streptokoky skupiny Viridans
Gram-negativní bakterie
Citrobacter koseri Escherichia coli Klebsiella pneumoniae Proteus mirabilis
Metody testování citlivosti
Je-li to možné, měla by klinická mikrobiologická laboratoř poskytovat pravidelné zprávy, které popisují regionální/lokální profil citlivosti potenciálních nozokomiálních a komunitních patogenů. Tyto zprávy by měly pomoci lékaři při výběru antibakteriálního léku pro léčbu.
Techniky ředění
Kvantitativní metody se používají ke stanovení antimikrobiálních minimálních inhibičních koncentrací (MIC). Tyto MIC poskytují odhady citlivosti bakterií na antimikrobiální sloučeniny. MIC by měly být stanoveny pomocí standardizované testovací metody1 (bujón a/nebo agar). Hodnoty MIC by měly být interpretovány podle kritérií uvedených v tabulce 1.
Difúzní techniky
Kvantitativní metody, které vyžadují měření průměrů zón, také poskytují reprodukovatelné odhady citlivosti bakterií na antimikrobiální sloučeniny. Velikost zóny by měla být stanovena pomocí standardizované metody.2 Postup využívá papírové disky napuštěné 5 mcg cefdiniru k testování citlivosti bakterií. Kritéria interpretace diskové difúze jsou uvedena v tabulce 1.
Citlivost stafylokoků na cefdinir lze odvodit z testování penicilinu a cefoxitinu nebo oxacilinu. Stafylokoky citlivé na oxacilin (cefoxitin) lze považovat za citlivé na cefdinir.3
Zpráva „citlivé“ naznačuje, že antimikrobiální látka pravděpodobně inhibuje růst patogenu, pokud antimikrobiální sloučenina dosáhne koncentrací v místě infekce nezbytných k inhibici růstu patogenu. Zpráva „Střední“ znamená, že výsledek by měl být považován za nejednoznačný, a pokud mikroorganismus není plně citlivý na alternativní, klinicky proveditelné léky, test by se měl opakovat. Tato kategorie implikuje možnou klinickou použitelnost v místech těla, kde je léčivo fyziologicky koncentrované, nebo v situacích, kdy lze použít vysokou dávku léčiva. Tato kategorie také poskytuje nárazníkovou zónu, která zabraňuje malým nekontrolovaným technickým faktorům způsobit velké nesrovnalosti ve výkladu. Zpráva „Rezistentní“ naznačuje, že antimikrobiální látka pravděpodobně nebude inhibovat růst patogenu, pokud antimikrobiální sloučenina dosáhne koncentrací obvykle dosažitelných v místě infekce; měla by být zvolena jiná terapie.
Kontrola kvality
Standardizované postupy testování citlivosti vyžadují použití laboratorních kontrol pro sledování a zajištění přesnosti a přesnosti zásob a činidel používaných v testu a techniky jednotlivce provádějícího test.1,2,3 Standardní prášek cefdiniru by měl poskytovat následující rozsah hodnot MIC, jak je uvedeno v tabulce 2. Pro techniku difúze používající 5 mcg disk by měla být splněna kritéria v tabulce 2.
Klinické studie
Komunitně získaná bakteriální pneumonie
V kontrolované, dvojitě zaslepené studii u dospělých a dospívajících provedené v USA byl cefdinir BID srovnáván s cefaclorem 500 mg TID. Použitím přísné hodnotitelnosti a kritérií mikrobiologické/klinické odpovědi 6 až 14 dní po terapii byly získány následující míry klinického vyléčení, předpokládané míry mikrobiologické eradikace a statistické výsledky:
Americká komunitní studie pneumonie Cefdinir vs Cefaclor
Ve druhé kontrolované, zkoušejícím zaslepené studii u dospělých a dospívajících provedené primárně v Evropě byl cefdinir BID srovnáván s amoxicilinem/klavulanátem 500/125 mg TID. Za použití přísných kritérií hodnotitelnosti a klinické odpovědi 6 až 14 dní po terapii byly získány následující míry klinického vyléčení, předpokládané míry mikrobiologické eradikace a statistické výsledky:
Studie Pneumonie získaná v Evropském společenství Cefdinir vs amoxicilin/klavulanát
Streptokoková faryngitida/tonzilitida
Ve čtyřech kontrolovaných studiích provedených ve Spojených státech amerických byl cefdinir srovnáván s 10denním podáváním penicilinu u dospělých, dospívajících a dětských pacientů. Dvě studie (jedna u dospělých a dospívajících, druhá u pediatrických pacientů) porovnávaly 10denní podávání cefdiniru QD nebo BID s penicilinem 250 mg nebo 10 mg/kg QID. Za použití přísné hodnotitelnosti a kritérií mikrobiologické/klinické odpovědi 5 až 10 dní po terapii byly získány následující míry klinického vyléčení, míry mikrobiologické eradikace a statistické výsledky:
Studie faryngitidy/tonzilitidy Cefdinir (10 dní) vs Penicilin (10 dní)
Dvě studie (jedna u dospělých a dospívajících, druhá u pediatrických pacientů) porovnávaly 5denní podávání cefdiniru dvakrát denně a 10 dnů penicilinu 250 mg nebo 10 mg/kg QID. Použitím přísné hodnotitelnosti a kritérií mikrobiologické/klinické odpovědi 4 až 10 dní po terapii byly získány následující míry klinického vyléčení, míry mikrobiologické eradikace a statistické výsledky:
Studie faryngitidy/tonzilitidy Cefdinir (5 dní) vs Penicilin (10 dní)
REFERENCE
1. Institut klinických a laboratorních standardů (CLSI). Metody ředění Testy antimikrobiální citlivosti na bakterie, které rostou aerobně; Schválený standard – desáté vydání. CLSI dokument M07-A10 [2015], Clinical and Laboratory Standards Institute, 950 West Valley Road, Suite 2500, Wayne, Pennsylvania 19087, USA.
2. Institut klinických a laboratorních standardů (CLSI). Výkonové standardy pro antimikrobiální diskové difúzní testy citlivosti; Schválený standard – dvanácté vydání. CLSI dokument M02-A12 [2015], Clinical and Laboratory Standards Institute, 950 West Valley Road, Suite 2500, Wayne, Pennsylvania 19087, USA.
3. Institut klinických a laboratorních standardů (CLSI). Výkonové standardy pro testování antimikrobiální citlivosti; Dvacátý pátý informační doplněk, CLSI dokument M100-S25 [2015], Clinical and Laboratory Standards Institute, 950 West Valley Road, Suite 2500, Wayne, Pennsylvania 19087, USA.
4. Cockcroft DW, Gault MH. Predikce clearance kreatininu ze sérového kreatininu. Nephron 1976;16:31-41.
5. Schwartz GJ, Haycock GB, Edelmann CM, Spitzer A. Jednoduchý odhad rychlosti glomerulární filtrace u dětí odvozený z délky těla a plazmatického kreatininu. Pediatrie 1976;58:259-63.
6. Schwartz GJ, Feld LG, Langford DJ. Jednoduchý odhad rychlosti glomerulární filtrace u donošených dětí během prvního roku života. J Pediatrics 1984;104:849-54.
INFORMACE PRO PACIENTA
Pacienti by měli být poučeni, že antibakteriální léky včetně přípravku OMNICEF 300 mg by měly být používány pouze k léčbě bakteriálních infekcí. Neléčí virové infekce (např. nachlazení). Je-li OMNICEF 300 mg předepisován k léčbě bakteriální infekce, je třeba pacientům říci, že ačkoli je běžné, že se na začátku léčby cítí lépe, lék by měl být užíván přesně podle pokynů. Vynechání dávek nebo nedokončení celé terapie může (1) snížit účinnost okamžité léčby a (2) zvýšit pravděpodobnost, že si bakterie vyvinou rezistenci a nebudou v budoucnu léčitelné přípravkem OMNICEF 300 mg nebo jinými antibakteriálními léky.
Antacida obsahující hořčík nebo hliník narušují absorpci cefdiniru. Pokud je během léčby přípravkem OMNICEF 300 mg vyžadován tento typ antacida, měl by se přípravek OMNICEF užívat alespoň 2 hodiny před nebo po podání antacid.
Doplňky železa, včetně multivitaminů, které obsahují železo, narušují vstřebávání cefdiniru. Pokud jsou během léčby přípravkem OMNICEF 300 mg vyžadovány doplňky železa, měl by se přípravek OMNICEF užívat alespoň 2 hodiny před nebo po podání.
Kojenecká výživa obohacená železem významně neinterferuje s absorpcí cefdiniru. Proto lze OMNICEF 300 mg pro perorální suspenzi podávat s kojeneckou výživou obohacenou železem.
Diabetičtí pacienti a pečovatelé by si měli být vědomi toho, že perorální suspenze obsahuje 2,86 g sacharózy na čajovou lžičku.
Průjem je častým problémem způsobeným antibiotiky, který obvykle končí, když je antibiotikum vysazeno. Někdy po zahájení léčby antibiotiky se u pacientů může vyvinout vodnatá a krvavá stolice (s žaludečními křečemi a horečkou nebo bez nich) dokonce až dva nebo více měsíců po užití poslední dávky antibiotika. Pokud k tomu dojde, pacienti by měli co nejdříve kontaktovat svého lékaře.