Zyvox 600mg Linezolid Použití, vedlejší účinky a dávkování. Cena v internetové lékárně. Generické léky bez předpisu.
Co je Zyvox 600 mg a jak se používá?
Zyvox je lék na předpis používaný k léčbě příznaků bakteriálních infekcí, jako je zápal plic, kožní infekce a infekce, které jsou odolné vůči jiným antibiotikům. Zyvox 600 mg lze užívat samostatně nebo s jinými léky.
Zyvox patří do třídy léků nazývaných antibiotika, jiné; oxazolidinony.
Není známo, zda je přípravek Zyvox bezpečný a účinný u dětí.
Jaké jsou možné vedlejší účinky přípravku Zyvox?
Zyvox může způsobit závažné nežádoucí účinky včetně:
- kopřivka,
- potíže s dýcháním,
- otok v obličeji nebo krku,
- závažná kožní reakce,
- horečka,
- bolest krku,
- pálení očí,
- bolest kůže,
- červená nebo fialová kožní vyrážka s puchýři a olupováním,
- problémy se zrakem,
- změny ve vašem vidění,
- silná bolest žaludku,
- průjem, který je vodnatý nebo krvavý,
- záchvaty,
- pocení,
- pocit úzkosti nebo třes,
- míchání,
- halucinace,
- horečka,
- chvění,
- rychlý srdeční tep,
- svalová ztuhlost,
- cukání,
- ztráta koordinace,
- nevolnost,
- zvracení,
- průjem,
- neobvyklá bolest svalů,
- potíže s dýcháním,
- bolest břicha,
- nepravidelná srdeční frekvence,
- závrať,
- cítit zimu,
- pocit velké slabosti nebo únavy,
- zmatek,
- vředy v ústech,
- kožní vředy,
- snadná tvorba modřin,
- neobvyklé krvácení,
- bledá kůže,
- studené ruce a nohy,
- závratě a
- dušnost
Pokud máte některý z výše uvedených příznaků, okamžitě vyhledejte lékařskou pomoc.
Mezi nejčastější vedlejší účinky přípravku Zyvox patří:
- nevolnost,
- zvracení,
- průjem,
- mírná kožní vyrážka,
- anémie (nízký počet červených krvinek),
- bolest hlavy a
- závrať
Informujte lékaře, pokud máte jakýkoli nežádoucí účinek, který vás obtěžuje nebo který neustupuje.
To nejsou všechny možné vedlejší účinky přípravku Zyvox. Pro více informací se zeptejte svého lékaře nebo lékárníka.
Zavolejte svého lékaře o radu ohledně nežádoucích účinků. Nežádoucí účinky můžete hlásit úřadu FDA na čísle 1-800-FDA-1088.
POPIS
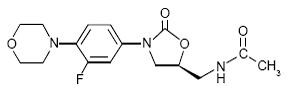
ZYVOX 600 mg IV injekce, ZYVOX tablety a ZYVOX pro perorální suspenzi obsahují linezolid, což je syntetická antibakteriální látka ze třídy oxazolidinonů. Chemický název linezolidu je (S)-N-[[3-[3-fluor-4-(4morfolinyl)fenyl]-2-oxo-5-oxazolidinyl]methyl]acetamid.
Empirický vzorec je C16H20FN3O4. Jeho molekulová hmotnost je 337,35 a jeho chemická struktura je znázorněna níže:
ZYVOX 600 mg IV injekce se dodává jako sterilní izotonický roztok připravený k použití pro intravenózní infuzi. Každý ml obsahuje 2 mg linezolidu. Neaktivní složky jsou citrát sodný, kyselina citrónová a dextróza ve vodném vehikulu pro intravenózní podání. Obsah sodíku (Na+) je 0,38 mg/ml (5 mEq/300 ml sáček a 1,7 mEq/100 ml sáček).
ZYVOX Tablet pro perorální podání obsahuje 600 mg linezolidu jako potahované lisované tablety. Neaktivní složky jsou kukuřičný škrob, mikrokrystalická celulóza, hydroxypropylcelulóza, sodná sůl glykolátu škrobu, stearát hořečnatý, hypromelóza, polyethylenglykol, oxid titaničitý a karnaubský vosk. Obsah sodíku (Na+) je 2,92 mg na 600mg tabletu (0,1 mEq/tabletu).
ZYVOX 600 mg pro perorální suspenzi se dodává jako granule/prášek s pomerančovou příchutí pro rekonstituci do suspenze pro perorální podání. Po rekonstituci obsahuje každých 5 ml 100 mg linezolidu. Neaktivní složky jsou sacharóza, kyselina citrónová, citrát sodný, mikrokrystalická celulóza a sodná sůl karboxymethylcelulózy, aspartam, xantanová guma, mannitol, benzoát sodný, koloidní oxid křemičitý, chlorid sodný a příchutě [viz INFORMACE PRO PACIENTA ]. Obsah sodíku (Na+) je 8,52 mg/5 ml (0,4 mEq/5 ml).
INDIKACE
Nozokomiální pneumonie
ZYVOX je indikován k léčbě nozokomiálních pneumonií způsobených Staphylococcus aureus (izoláty citlivé a rezistentní na meticilin) nebo Streptococcus pneumoniae [viz Klinické studie ].
Pneumonie získaná v komunitě
ZYVOX 600 mg je indikován k léčbě komunitní pneumonie způsobené Streptococcus pneumoniae, včetně případů se současnou bakteriémií nebo Staphylococcus aureus (pouze izoláty citlivé na meticilin) [viz Klinické studie ].
Komplikované infekce kůže a kožní struktury
ZYVOX 600 mg je indikován k léčbě komplikovaných infekcí kůže a kožní struktury, včetně infekcí diabetické nohy, bez souběžné osteomyelitidy, způsobené Staphylococcus aureus (izoláty citlivé a rezistentní na meticilin), Streptococcus pyogenes nebo Streptococcus agalactiae. ZYVOX nebyl studován v léčbě dekubitálních vředů [viz Klinické studie ].
Nekomplikované infekce kůže a kožní struktury
ZYVOX 600 mg je indikován k léčbě nekomplikovaných infekcí kůže a kožních struktur způsobených Staphylococcus aureus (pouze izoláty citlivé na meticilin) nebo Streptococcus pyogenes [viz Klinické studie ].
Infekce Enterococcus faecium rezistentní na vankomycin
ZYVOX 600 mg je indikován k léčbě infekcí Enterococcus faecium rezistentních na vankomycin, včetně případů se současnou bakteriémií [viz Klinické studie ].
Omezení použití
- ZYVOX 600 mg není indikován k léčbě gramnegativních infekcí. Je důležité, aby byla okamžitě zahájena specifická gramnegativní terapie, pokud je zdokumentován nebo existuje podezření na doprovodný gramnegativní patogen [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
- Bezpečnost a účinnost přípravků ZYVOX 600 mg podávaných déle než 28 dnů nebyla hodnocena v kontrolovaných klinických studiích (viz Klinické studie ].
Používání
Aby se omezil vývoj bakterií rezistentních vůči lékům a zachovala se účinnost ZYVOX 600 mg a dalších antibakteriálních léků, měl by být ZYVOX používán pouze k léčbě nebo prevenci infekcí, u kterých je prokázáno nebo existuje silné podezření, že jsou způsobeny citlivými bakteriemi. Pokud jsou k dispozici informace o kultivaci a citlivosti, je třeba je vzít v úvahu při výběru nebo úpravě antibakteriální terapie. Při absenci takových údajů mohou k empirickému výběru terapie přispět místní epidemiologie a vzorce citlivosti.
DÁVKOVÁNÍ A PODÁVÁNÍ
Obecné dávkování a podávání
Doporučené dávkování pro přípravky ZYVOX 600 mg pro léčbu infekcí je popsáno v tabulce 1.
Při přechodu z intravenózního na perorální podávání není nutná žádná úprava dávky.
Intravenózní podání
ZYVOX 600 mg IV injekce se dodává v jednodávkových infuzních vacích připravených k použití. Parenterální léčivé přípravky by měly být před podáním vizuálně zkontrolovány na přítomnost částic. Pevným stlačením sáčku zkontrolujte, zda nedochází k nepatrným únikům. Pokud zjistíte netěsnosti, roztok zlikvidujte, protože může být narušena sterilita. Uchovávejte infuzní sáčky v přebalu, dokud nejsou připraveny k použití. Skladujte při pokojové teplotě. Chraňte před mrazem. ZYVOX 600 mg IV injekce může vykazovat žluté zbarvení, které může časem zesílit bez nepříznivého ovlivnění účinnosti.
ZYVOX IV Injection by měl být podáván intravenózní infuzí po dobu 30 až 120 minut. Nepoužívejte tento intravenózní infuzní vak v sériovém zapojení. Do tohoto roztoku by se neměly přidávat přísady. Pokud má být ZYVOX 600 mg IV injekce podáván současně s jiným lékem, každý lék by měl být podáván samostatně v souladu s doporučeným dávkováním a způsobem podání pro každý přípravek. Nepoužitou část zlikvidujte.
Pokud se stejná intravenózní linka používá pro sekvenční infuzi několika léků, je třeba tuto linku propláchnout před a po infuzi ZYVOX IV injekce infuzním roztokem kompatibilním se ZYVOX 600 mg IV injekcí a jakýmkoli jiným lékem podávaným prostřednictvím této společné linky .
Kompatibilita
Kompatibilní intravenózní roztoky zahrnují 0,9% injekci chloridu sodného, USP, 5% injekci dextrózy, USP a laktátovou Ringerovu injekci, USP.
Nekompatibility
Fyzikální nekompatibilita nastala, když byl ZYVOX 600 mg IV injekce kombinován s následujícími léky během simulovaného podání do Y: amfotericin B, chlorpromazin HCl, diazepam, pentamidin isothionát, erythromycin laktobionát, fenytoin sodný a trimethoprim-sulfamethoxazol. Navíc došlo k chemické inkompatibilitě, když byl ZYVOX IV Injection kombinován s ceftriaxonem sodným.
Konstituce ústního pozastavení
ZYVOX pro perorální suspenzi se dodává jako prášek/granule pro rekonstituci. Jemně poklepejte na lahvičku, aby se prášek uvolnil. Přidejte celkem 123 ml destilované vody ve dvou dávkách. Po přidání první poloviny důkladně protřepejte, aby se všechen prášek smočil. Poté přidejte druhou polovinu vody a důkladně protřepejte, abyste získali stejnoměrnou suspenzi. Po rekonstituci obsahuje každých 5 ml suspenze 100 mg linezolidu. Před použitím jemně promíchejte převrácením lahvičky 3 až 5 krát. Netřepat. Připravenou suspenzi skladujte při pokojové teplotě. Spotřebujte do 21 dnů po konstituci.
JAK DODÁVÁNO
Dávkové formy A Síly
ZYVOX 600 mg IV injekce: 200 mg/100 ml (2 mg/ml) a 600 mg/300 ml (2 mg/ml) linezolid, jednorázové flexibilní plastové infuzní vaky připravené k použití v přebalu z laminátové fólie.
ZYVOX 600 mg tablety:
bílá potahovaná tableta ve tvaru tobolky s vyraženým „ZYV“ na jedné straně a „600“ na druhé straně ZYVOX 600 mg pro perorální suspenzi: suché, bílé až téměř bílé granule/prášek s pomerančovou příchutí. Po složení podle návodu bude každá lahvička obsahovat 150 ml suspenze poskytující ekvivalent 100 mg linezolidu na každých 5 ml.
Skladování A Manipulace
Injekce
ZYVOX 600 mg IV Injekce je k dispozici v jednodávkových flexibilních plastových infuzních vacích připravených k použití ve fóliovém laminátovém přebalu. Infuzní vaky a porty nejsou vyrobeny z přírodního kaučukového latexu. Infuzní sáčky jsou dostupné v následujících velikostech balení:
200 mg/100 ml (2 mg/ml) linezolid x 10 - NDC 0009-5137-04 600 mg/300 ml (2 mg/ml) linezolid x 10 - NDC 0009-5140-04
Tablety
ZYVOX 600 mg tablety jsou k dispozici následovně:
600 mg (bílé potahované tablety ve tvaru tobolky s vyraženým „ZYV“ na jedné straně a „600“ na druhé straně)
20 tablet v HDPE lahvičce - NDC 0009-5138-02 Jednodávkové balení po 30 tabletách - NDC 0009-5138-03
Orální suspenze
ZYVOX pro perorální suspenzi je k dispozici jako suché bílé až téměř bílé granule/prášek s pomerančovou příchutí. Po složení podle návodu bude každá lahvička obsahovat 150 ml suspenze poskytující ekvivalent 100 mg linezolidu na každých 5 ml. ZYVOX pro perorální suspenzi se dodává následovně:
100 mg/5 ml ve skleněných lahvičkách o objemu 240 ml - NDC 0009-5136-01
Skladování A Manipulace
Uchovávejte při teplotě 25 °C (77 °F). Chraňte před světlem. Uchovávejte lahvičky dobře uzavřené, aby byly chráněny před vlhkostí. Infuzní vaky se doporučuje uchovávat v přebalu, dokud nejsou připraveny k použití. Chraňte infuzní sáčky před mrazem.
Distribuováno divizí Pharmacia & Upjohn Co Division společnosti Pfizer Inc, NY, NY 10017. Revize: listopad 2021.
VEDLEJŠÍ EFEKTY
Zkušenosti z klinických studií
Vzhledem k tomu, že klinické studie jsou prováděny za velmi odlišných podmínek, nelze míry nežádoucích reakcí pozorované v klinických studiích léku přímo srovnávat s mírami v klinických studiích jiného léku a nemusí odrážet míry pozorované v praxi.
Dospělí
Bezpečnost přípravků ZYVOX 600 mg byla hodnocena u 2 046 dospělých pacientů zařazených do sedmi klinických studií fáze 3 kontrolovaných komparátorem, kteří byli léčeni po dobu až 28 dnů.
pacientů léčených pro nekomplikované infekce kůže a kožních struktur (uSSSI) se u 25,4 % pacientů léčených přípravkem ZYVOX a 19,6 % pacientů léčených komparátorem vyskytl alespoň jeden nežádoucí účinek související s lékem. U všech ostatních indikací se u 20,4 % pacientů léčených přípravkem ZYVOX a 14,3 % pacientů léčených komparátorem vyskytl alespoň jeden nežádoucí účinek související s lékem.
Tabulka 2 ukazuje výskyt nežádoucích účinků souvisejících se všemi příčinami, které se objevily při léčbě, hlášených u nejméně 1 % dospělých pacientů v těchto studiích podle dávky ZYVOXu.
Z pacientů léčených pro uSSSI přerušilo léčbu 3,5 % pacientů léčených ZYVOXem a 2,4 % pacientů léčených komparátorem kvůli nežádoucím účinkům souvisejícím s lékem. U všech ostatních indikací došlo k přerušení léčby z důvodu nežádoucích účinků souvisejících s lékem u 2,1 % pacientů léčených přípravkem ZYVOX a 1,7 % pacientů léčených komparátorem. Nejčastějšími hlášenými nežádoucími účinky souvisejícími s lékem, které vedly k přerušení léčby, byly nauzea, bolest hlavy, průjem a zvracení.
Pediatričtí pacienti
Bezpečnost přípravků ZYVOX byla hodnocena u 215 dětských pacientů ve věku od narození do 11 let au 248 dětských pacientů ve věku 5 až 17 let (146 z těchto 248 bylo ve věku 5 až 11 let a 102 bylo ve věku 12 až 17 let). Tito pacienti byli zařazeni do dvou klinických studií fáze 3 kontrolovaných komparátorem a byli léčeni po dobu až 28 dnů. Ve studii hospitalizovaných pediatrických pacientů (od narození do 11 let) s grampozitivními infekcemi, kteří byli randomizováni 2 ku 1 (linezolid: vankomycin), byla mortalita 6,0 % (13/215) v rameni s linezolidem a 3,0 % (3/101) v rameni vankomycinu. Vzhledem k závažnému základnímu onemocnění v populaci pacientů však nebylo možné prokázat žádnou kauzalitu.
Z pediatrických pacientů léčených pro uSSSI se u 19,2 % pacientů léčených přípravkem ZYVOX a 14,1 % pacientů léčených komparátorem vyskytl alespoň jeden nežádoucí účinek související s lékem. U všech ostatních indikací se u 18,8 % pacientů léčených přípravkem ZYVOX a 34,3 % pacientů léčených komparátorem vyskytl alespoň jeden nežádoucí účinek související s lékem.
Tabulka 3 ukazuje výskyt nežádoucích účinků souvisejících se všemi příčinami a souvisejících s léčbou hlášených u více než 1 % pediatrických pacientů (a více než 1 pacienta) v obou léčebných skupinách ve studiích fáze 3 kontrolovaných komparátorem.
Z pediatrických pacientů léčených pro uSSSI přerušilo léčbu 1,6 % pacientů léčených přípravkem ZYVOX a 2,4 % pacientů léčených komparátorem kvůli nežádoucím účinkům souvisejícím s lékem. U všech ostatních indikací došlo k přerušení léčby z důvodu nežádoucích účinků souvisejících s lékem u 0,9 % pacientů léčených přípravkem ZYVOX a 6,1 % pacientů léčených komparátorem.
Laboratorní abnormality
ZYVOX byl spojován s trombocytopenií, pokud se používal v dávkách až 600 mg včetně každých 12 hodin po dobu až 28 dnů. Ve 3. fázi kontrolovaných srovnávacích studií bylo procento dospělých pacientů, u kterých se vyvinul podstatně nízký počet krevních destiček (definovaný jako méně než 75 % spodní hranice normálu a/nebo výchozí hodnoty) 2,4 % (rozmezí mezi studiemi: 0,3 až 10,0 %) se ZYVOX 600 mg a 1,5 % (rozmezí mezi studiemi: 0,4 až 7,0 %) s komparátorem. Ve studii hospitalizovaných pediatrických pacientů ve věku od narození do 11 let bylo procento pacientů, u kterých se vyvinul podstatně nízký počet krevních destiček (definovaný jako méně než 75 % spodní hranice normálu a/nebo výchozí hodnoty), 12,9 % při ZYVOX 600 mg a 13,4 % s vankomycinem. V ambulantní studii u pediatrických pacientů ve věku od 5 do 17 let bylo procento pacientů, u kterých se vyvinul podstatně nízký počet krevních destiček, 0 % u přípravku ZYVOX a 0,4 % u cefadroxilu. Zdá se, že trombocytopenie spojená s užíváním přípravku ZYVOX závisí na délce léčby (obecně delší než 2 týdny léčby). Počet krevních destiček se u většiny pacientů vrátil do normálního rozmezí/výchozí hodnoty během období sledování. V klinických studiích fáze 3 nebyly u pacientů, u kterých se rozvinula trombocytopenie, identifikovány žádné související klinické nežádoucí účinky. U pacientů s trombocytopenií byly v programu použití přípravku ZYVOX ze soucitu zjištěny příhody krvácení; roli linezolidu v těchto příhodách nelze určit [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Změny pozorované v ostatních laboratorních parametrech, bez ohledu na lékový vztah, neodhalily žádné podstatné rozdíly mezi ZYVOX 600 mg a srovnávacími látkami. Tyto změny nebyly obecně klinicky významné, nevedly k přerušení léčby a byly reverzibilní. Výskyt dospělých a dětských pacientů s alespoň jednou v podstatě abnormální hematologickou nebo chemickou hodnotou séra je uveden v tabulkách 4, 5, 6 a 7.
Postmarketingové zkušenosti
Následující nežádoucí účinky byly zjištěny během používání přípravku ZYVOX po schválení. Protože jsou tyto reakce hlášeny dobrovolně z populace nejisté velikosti, není vždy možné spolehlivě odhadnout jejich frekvenci nebo stanovit příčinnou souvislost s expozicí léku:
- Myelosuprese (včetně anémie, leukopenie, pancytopenie a trombocytopenie) (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]; sideroblastická anémie.
- Periferní neuropatie a neuropatie zrakového nervu někdy progredující ke ztrátě zraku [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
- Laktátová acidóza [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ]. Ačkoli se tato hlášení primárně vyskytovala u pacientů léčených déle, než je maximální doporučená délka 28 dnů, byly tyto příhody hlášeny také u pacientů, kteří dostávali kratší léčebné cykly.
- Serotoninový syndrom byl hlášen u pacientů užívajících současně serotonergní látky, včetně antidepresiv, jako jsou selektivní inhibitory zpětného vychytávání serotoninu (SSRI) a opioidy, a ZYVOX (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
- Křeče [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
- Anafylaxe, angioedém, bulózní kožní poruchy včetně závažných kožních nežádoucích reakcí (SCAR), jako je toxická epidermální nekrolýza a Stevens-Johnsonův syndrom, a hypersenzitivní vaskulitida.
- Při užívání linezolidu bylo hlášeno povrchové zbarvení zubů a zbarvení jazyka. Zbarvení zubu bylo v případech se známým výsledkem odstranitelné profesionálním čištěním zubů (ruční odstranění vodního kamene).
- Hypoglykémie, včetně symptomatických epizod [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
- Hyponatremie a/nebo syndrom nepřiměřené sekrece antidiuretického hormonu (SIADH) [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
DROGOVÉ INTERAKCE
Inhibitory monoaminoxidázy
Linezolid je reverzibilní, neselektivní inhibitor monoaminooxidázy [viz KONTRAINDIKACE a KLINICKÁ FARMAKOLOGIE ].
Adrenergní a serotonergní látky
Linezolid má potenciál pro interakci s adrenergními a serotonergními látkami [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ a KLINICKÁ FARMAKOLOGIE ].
VAROVÁNÍ
Zahrnuto jako součást "OPATŘENÍ" Sekce
OPATŘENÍ
Myelosuprese
pacientů užívajících linezolid byla hlášena myelosuprese (včetně anémie, leukopenie, pancytopenie a trombocytopenie). V případech, kdy je výsledek znám, se po vysazení linezolidu zvýšily ovlivněné hematologické parametry na úroveň před léčbou. U pacientů, kteří užívají linezolid, zejména u pacientů, kteří užívají linezolid déle než dva týdny, u pacientů s již existující myelosupresí, u pacientů, kteří současně užívají léky způsobující útlum kostní dřeně nebo u pacientů s chronickou infekcí, by měl být každý týden sledován kompletní krevní obraz. užívali předchozí nebo souběžnou léčbu antibakteriálními léky. U pacientů, u kterých se rozvine nebo se zhorší myelosuprese, je třeba zvážit přerušení léčby linezolidem.
Periferní a optická neuropatie
pacientů léčených přípravkem ZYVOX 600 mg byly hlášeny periferní a optické neuropatie, především u pacientů léčených déle, než je maximální doporučená délka 28 dnů. V případech neuropatie zrakového nervu, která progredovala do ztráty zraku, byli pacienti léčeni po delší dobu, než je maximální doporučená doba trvání. U některých pacientů léčených přípravkem ZYVOX po dobu kratší než 28 dní bylo hlášeno rozmazání zraku. U dětí byla také hlášena periferní a optická neuropatie.
Pokud se u pacientů objeví příznaky poškození zraku, jako jsou změny zrakové ostrosti, změny barevného vidění, rozmazané vidění nebo porucha zorného pole, doporučuje se rychlé oftalmologické vyšetření. Zrakové funkce by měly být sledovány u všech pacientů užívajících ZYVOX po delší dobu (≥ 3 měsíce) a u všech pacientů, kteří hlásili nové zrakové symptomy bez ohledu na délku léčby přípravkem ZYVOX. Pokud se objeví periferní nebo optická neuropatie, je třeba zvážit další užívání přípravku ZYVOX 600 mg u těchto pacientů s ohledem na možná rizika.
serotoninový syndrom
Byly hlášeny spontánní zprávy o serotoninovém syndromu včetně fatálních případů spojených se současným podáváním ZYVOXu a serotonergních látek, včetně antidepresiv, jako jsou selektivní inhibitory zpětného vychytávání serotoninu (SSRI).
Pokud to není klinicky vhodné a pacienti nejsou pečlivě sledováni z hlediska známek a/nebo příznaků serotoninového syndromu nebo reakcí podobných neuroleptickému malignímu syndromu (NMS), neměl by být linezolid podáván pacientům s karcinoidovým syndromem a/nebo pacientům užívajícím některý z následujících léků : inhibitory zpětného vychytávání serotoninu, tricyklická antidepresiva, bupropion, buspiron, agonisté serotoninového 5-HT1 receptoru (triptany) a opioidy, včetně meperidinu [viz DROGOVÉ INTERAKCE a KLINICKÁ FARMAKOLOGIE ].
některých případech může pacient, který již dostává serotonergní antidepresivum nebo buspiron, vyžadovat urgentní léčbu linezolidem. Pokud alternativy k linezolidu nejsou k dispozici a potenciální přínosy linezolidu převažují nad riziky serotoninového syndromu nebo reakcí podobných NMS, serotonergní antidepresivum by mělo být okamžitě ukončeno a linezolid by měl být podán. Pacient by měl být sledován po dobu dvou týdnů (pět týdnů, pokud byl užíván fluoxetin) nebo do 24 hodin po poslední dávce linezolidu, podle toho, co nastane dříve. Příznaky serotoninového syndromu nebo reakce podobné NMS zahrnují hypertermii, rigiditu, myoklonus, autonomní nestabilitu a změny duševního stavu, které zahrnují extrémní neklid přecházející do deliria a kómatu. Pacient by měl být také sledován kvůli symptomům z vysazení antidepresiva (popis souvisejících symptomů z vysazení viz příbalový leták konkrétního léku(ů)).
Nerovnováha úmrtnosti ve vyšetřovací studii u pacientů s infekcemi krevního řečiště souvisejícími s katétrem, včetně pacientů s infekcemi v místě katétru
Nerovnováha v úmrtnosti byla pozorována u pacientů léčených linezolidem ve srovnání s vankomycinem/dicloxacilinem/oxacilinem v otevřené studii u vážně nemocných pacientů s infekcemi souvisejícími s intravaskulárním katetrem [78/363 (21,5 %) vs. 58/363 (16,0 % ); poměr šancí 1,426, 95% CI 0,970, 2,098]. I když kauzalita nebyla stanovena, tato pozorovaná nerovnováha se vyskytla především u pacientů léčených linezolidem, u kterých byly na začátku identifikovány buď gramnegativní patogeny, smíšené gramnegativní a grampozitivní patogeny nebo žádný patogen, ale nebyla pozorována u pacientů s Pouze grampozitivní infekce.
Linezolid není schválen a neměl by se používat k léčbě pacientů s infekcemi krevního řečiště souvisejícími s katétrem nebo infekcemi v místě katétru.
Linezolid nemá klinickou aktivitu proti gramnegativním patogenům a není indikován k léčbě gramnegativních infekcí. Je důležité, aby byla okamžitě zahájena specifická gramnegativní terapie, pokud je zdokumentován nebo existuje podezření na doprovodný gramnegativní patogen [viz INDIKACE ].
Průjem spojený s Clostridioides Difficile
Průjem spojený s Clostridioides difficile (CDAD) byl hlášen při použití téměř všech antibakteriálních látek, včetně ZYVOX 600 mg, a může se pohybovat v rozsahu od mírného průjmu až po fatální kolitidu. Léčba antibakteriálními látkami mění normální flóru tlustého střeva, což vede k přemnožení C. difficile.
C. difficile produkuje toxiny A a B, které přispívají k rozvoji CDAD. Kmeny C. difficile produkující hypertoxin způsobují zvýšenou morbiditu a mortalitu, protože tyto infekce mohou být odolné vůči antimikrobiální léčbě a mohou vyžadovat kolektomii. CDAD je třeba vzít v úvahu u všech pacientů, kteří mají průjem po užití antibakteriálních léků.
Je nutná pečlivá anamnéza, protože CDAD se objevilo více než dva měsíce po podání antibakteriálních látek.
Pokud je podezření na CDAD nebo je potvrzeno, může být nutné přerušit pokračující užívání antibakteriálních léků, které nejsou zaměřeny proti C. difficile. Podle klinické indikace by měla být zahájena vhodná léčba tekutin a elektrolytů, suplementace proteinů, antibakteriální léčba C. difficile a chirurgické vyšetření.
Potenciální interakce způsobující zvýšení krevního tlaku
Pokud nejsou pacienti sledováni kvůli možnému zvýšení krevního tlaku, neměl by být linezolid podáván pacientům s nekontrolovanou hypertenzí, feochromocytomem, tyreotoxikózou a/nebo pacientům užívajícím některý z následujících typů léků: přímo a nepřímo působící sympatomimetika (např. pseudoefedrin), vazopresivní látky (např. adrenalin, norepinefrin), dopaminergní látky (např. dopamin, dobutamin) [viz DROGOVÉ INTERAKCE a KLINICKÁ FARMAKOLOGIE ].
Laktátová acidóza
Při užívání přípravku ZYVOX byla hlášena laktátová acidóza. V hlášených případech se u pacientů vyskytly opakované epizody nevolnosti a zvracení. Pacienti, u kterých se během léčby přípravkem ZYVOX objeví opakující se nevolnost nebo zvracení, nevysvětlitelná acidóza nebo nízká hladina bikarbonátu, by měli být okamžitě vyšetřeni lékařem.
Křeče
pacientů léčených linezolidem byly hlášeny křeče. V některých z těchto případů byly v anamnéze hlášeny záchvaty nebo rizikové faktory pro záchvaty.
Hypoglykémie
Postmarketingové případy symptomatické hypoglykemie byly hlášeny u pacientů s diabetes mellitus, kteří dostávali inzulín nebo perorální hypoglykemická léčiva při léčbě linezolidem, reverzibilním, neselektivním inhibitorem MAO. Některé inhibitory MAO byly spojovány s hypoglykemickými epizodami u diabetických pacientů užívajících inzulín nebo hypoglykemická činidla. Zatímco příčinná souvislost mezi linezolidem a hypoglykémií nebyla stanovena, diabetici by měli být při léčbě linezolidem varováni před možnými hypoglykemickými reakcemi.
Dojde-li k hypoglykemii, může být nutné snížit dávku inzulinu nebo perorálního hypoglykemika nebo vysadit perorální hypoglykemiku, inzulin nebo linezolid.
Hyponatremie a/nebo syndrom nepřiměřené sekrece antidiuretického hormonu (SIADH)
pacientů léčených linezolidem byly po uvedení na trh pozorovány případy hyponatremie a/nebo syndromu nepřiměřené sekrece antidiuretického hormonu (SIADH). V hlášených případech příznaky a symptomy zahrnovaly zmatenost, ospalost, všeobecnou slabost a v závažných případech vedly k selhání dýchání a dokonce ke smrti. Pravidelně sledujte hladiny sodíku v séru u starších osob, u pacientů užívajících diuretika a u dalších pacientů s rizikem hyponatremie a/nebo SIADH při užívání ZYVOXu. Pokud se objeví známky a příznaky hyponatremie a/nebo SIADH, přerušte léčbu přípravkem ZYVOX 600 mg a zaveďte vhodná podpůrná opatření.
Rizika u pacientů s fenylketonurií
Fenylalanin může být škodlivý pro pacienty s fenylketonurií (PKU). ZYVOX pro perorální suspenzi obsahuje fenylalanin, složku aspartamu. Každých 5 ml 100 mg/5 ml perorální suspenze obsahuje 20 mg fenylalaninu. Než předepíšete ZYVOX 600 mg pro perorální suspenzi pacientovi s PKU, zvažte kombinované denní množství fenylalaninu ze všech zdrojů, včetně ZYVOXu pro perorální suspenzi.
Ostatní přípravky ZYVOX neobsahují fenylalanin.
Vývoj bakterií odolných vůči léčivům
Je nepravděpodobné, že by předepsání přípravku ZYVOX při absenci prokázané nebo silně suspektní bakteriální infekce nebo profylaktické indikace přineslo pacientovi prospěch a zvýšilo riziko rozvoje bakterií rezistentních na léky.
Neklinická toxikologie
Karcinogeneze, mutageneze, zhoršení plodnosti
Celoživotní studie na zvířatech k hodnocení karcinogenního potenciálu linezolidu nebyly provedeny. Nebyl nalezen ani mutagenní, ani klastogenní potenciál v řadě testů, včetně: testů mutagenity (Amesova bakteriální reverze a mutace CHO buněk), testu in vitro neplánované syntézy DNA (UDS), testu in vitro chromozomových aberací v lidských lymfocytech a testu in vivo myší mikronukleový test.
Linezolid neovlivnil fertilitu ani reprodukční schopnost dospělých potkaních samic, kterým byly perorálně podávány dávky až 100 mg/kg/den po dobu 14 dnů před pářením do 7. dne březosti. dávky ≥ 50 mg/kg/den, s expozicemi přibližně stejnými nebo vyššími, než je očekávaná hladina expozice u člověka (srovnání expozice jsou založena na AUC). Reverzibilní účinky na fertilitu byly zprostředkovány prostřednictvím změněné spermatogeneze. Postižené spermatidy obsahovaly abnormálně vytvořené a orientované mitochondrie a byly neživotaschopné. Hypertrofie a hyperplazie epiteliálních buněk v nadvarleti byla pozorována ve spojení se sníženou plodností. Podobné změny nadvarlete nebyly pozorovány u psů.
pohlavně dospělých samců potkanů, kteří byli vystaveni léku jako juvenilní jedinci, byla po léčbě linezolidem pozorována mírně snížená plodnost po většinu období jejich sexuálního vývoje (50 mg/kg/den od 7. do 36. dne věku a 100 mg/kg/den od 37. do 55. dne věku), s expozicemi až 1,7krát vyššími, než jsou průměrné hodnoty AUC pozorované u pediatrických pacientů ve věku od 3 měsíců do 11 let. Snížená plodnost nebyla pozorována při kratších léčebných obdobích, což odpovídá expozici in utero v časném novorozeneckém období (6. den těhotenství až 5. den po narození), expozici novorozenců (5. až 21. den po narození) nebo expozici mladistvých (22. až 35. den po narození) ). U potkanů léčených od 22. do 35. dne po narození bylo pozorováno reverzibilní snížení motility spermií a změněná morfologie spermií.
Použití u konkrétních populací
Těhotenství
Shrnutí rizik
Dostupné údaje z publikovaných a postmarketingových kazuistik s užíváním linezolidu u těhotných žen neidentifikovaly s lékem spojené riziko závažných vrozených vad, potratu nebo nepříznivých mateřských nebo fetálních výsledků. Když byl linezolid podáván během organogeneze, nezpůsobil malformace u myší, potkanů nebo králíků při hladinách mateřské expozice přibližně 6,5násobku (myši), ekvivalentním (potkani) nebo 0,06násobku (králíci) klinické terapeutické expozici na základě AUC. Avšak embryo-fetální letalita byla pozorována u myší při 6,5násobku odhadované expozice u člověka. Když byly samičkám potkanů podávány dávky během organogeneze během laktace, postnatální přežití mláďat se snížilo v dávkách přibližně ekvivalentních odhadované expozici u člověka na základě AUC (viz Data ).
Základní riziko závažných vrozených vad a potratu pro uvedené populace není známo. Všechna těhotenství mají na pozadí riziko vrozené vady, ztráty nebo jiných nepříznivých následků. V obecné populaci USA je odhadované základní riziko závažných vrozených vad a potratu u klinicky uznaných těhotenství 2–4 % a 15–20 %.
Data
Údaje o zvířatech
U myší byla embryo-fetální toxicita pozorována pouze při dávkách, které způsobily toxicitu pro matku (klinické příznaky a snížený přírůstek tělesné hmotnosti). Perorální dávka 450 mg/kg/den podaná ode dne gestace (GD) 6-16 (6,5násobek odhadované expozice u člověka na základě AUC) korelovala se zvýšenou postimplantační smrtí embryí, včetně celkové ztráty vrhu, snížení tělesné hmotnosti plodu a zvýšený výskyt fúze žeberní chrupavky. Při dávkách do 150 mg/kg/den nebyla pozorována toxicita pro matku ani embryo-fetální toxicitu. Fetální malformace nebyly pozorovány.
potkanů byla pozorována fetální toxicita při dávkách 15 a 50 mg/kg/den podaných perorálně od GD 6-17 (expozice 0,22krát přibližně ekvivalentní odhadované expozici u člověka na základě AUC). Účinky spočívaly ve snížení tělesné hmotnosti plodu a snížení osifikace hrudních kostí, což je nález často pozorovaný ve spojení se sníženou tělesnou hmotností plodu. Fetální malformace nebyly pozorovány. Mateřská toxicita ve formě sníženého přírůstku tělesné hmotnosti byla pozorována při dávce 50 mg/kg/den.
U králíků se snížení tělesné hmotnosti plodu objevilo pouze v přítomnosti mateřské toxicity (klinické příznaky, snížený přírůstek tělesné hmotnosti a spotřeba potravy), když byla podávána v perorální dávce 15 mg/kg/den podávané od 6-20 GD (0,06násobek odhadovaná expozice u člověka na základě AUC). Fetální malformace nebyly pozorovány.
Když byly samičkám potkanů během březosti a laktace (GD 6 až 20. den laktace) podáváno 50 mg/kg/den (přibližně ekvivalentní odhadované expozici u člověka na základě AUC) linezolidu, přežívání mláďat se v postnatálních dnech 1 až 4 snížilo Samci a samice, kterým bylo umožněno dospět do reprodukčního věku, když se spářili, vykazovali zvýšení preimplantačních ztrát.
Laktace
Shrnutí rizik
Linezolid je přítomen v mateřském mléce. Na základě údajů z dostupných publikovaných kazuistik by denní dávka linezolidu, kterou by dítě dostávalo z mateřského mléka, byla přibližně 6 % až 9 % doporučené terapeutické dávky kojence (10 mg/kg každých 8 hodin). Neexistují žádné informace o účincích linezolidu na kojené dítě; průjem a zvracení však byly nejčastějšími nežádoucími účinky hlášenými v klinických studiích u kojenců užívajících linezolid terapeuticky (viz NEŽÁDOUCÍ REAKCE a (viz Klinické úvahy). Neexistují žádné informace o účincích linezolidu na produkci mléka. Je třeba zvážit vývojové a zdravotní přínosy kojení spolu s klinickou potřebou matky užívat linezolid a veškerými potenciálními nežádoucími účinky linezolidu nebo základního onemocnění matky na kojené dítě.
Klinické úvahy
Doporučte kojícím ženám, aby sledovaly kojené dítě kvůli průjmu a zvracení.
Ženy a muži reprodukčního potenciálu
Neplodnost
Muži
Na základě zjištění ze studií na potkanech může ZYVOX reverzibilně narušit plodnost u pacientů mužského pohlaví (viz Neklinická toxikologie )].
Pediatrické použití
Bezpečnost a účinnost přípravku ZYVOX 600 mg pro léčbu pediatrických pacientů s následujícími infekcemi je podpořena důkazy z adekvátních a dobře kontrolovaných studií u dospělých, farmakokinetickými údaji u pediatrických pacientů a dalšími údaji z komparátorem kontrolované studie grampozitivních infekce u dětských pacientů ve věku od narození do 11 let [viz INDIKACE , KLINICKÁ FARMAKOLOGIE a Klinické studie ]:
- nozokomiální pneumonie
- komplikované infekce kůže a kožní struktury
- komunitní pneumonie (také podpořená důkazy z nekontrolované studie u pacientů ve věku od 8 měsíců do 12 let)
- infekce Enterococcus faecium rezistentní na vankomycin
Bezpečnost a účinnost přípravku ZYVOX 600 mg pro léčbu pediatrických pacientů s následující infekcí byla stanovena ve srovnávací kontrolované studii u pediatrických pacientů ve věku od 5 do 17 let [viz Klinické studie ]:
- nekomplikované infekce kůže a kožní struktury způsobené Staphylococcus aureus (pouze kmeny citlivé na meticilin) nebo Streptococcus pyogenes
Farmakokinetické informace získané u pediatrických pacientů s ventrikuloperitoneálními zkraty ukázaly proměnlivé koncentrace linezolidu v mozkomíšním moku (CSF) po jednorázovém a opakovaném podání linezolidu; terapeutické koncentrace nebyly v CSF konzistentně dosahovány nebo udržovány. Proto se použití linezolidu k empirické léčbě pediatrických pacientů s infekcemi centrálního nervového systému nedoporučuje.
Farmakokinetika linezolidu byla hodnocena u pediatrických pacientů od narození do 17 let věku. Obecně se clearance linezolidu v závislosti na hmotnosti postupně snižuje s rostoucím věkem pediatrických pacientů. U předčasně narozených (gestační věk DÁVKOVÁNÍ A PODÁVÁNÍ a KLINICKÁ FARMAKOLOGIE ].
Podle omezené klinické zkušenosti bylo 5 ze 6 (83 %) pediatrických pacientů s infekcemi způsobenými grampozitivními patogeny s minimálními inhibičními koncentracemi (MIC) 4 mcg/ml léčených přípravkem ZYVOX 600 mg klinicky vyléčeno. Pediatričtí pacienti však vykazují širší variabilitu clearance linezolidu a systémové expozice (AUC) ve srovnání s dospělými. U pediatrických pacientů se suboptimální klinickou odpovědí, zejména u pacientů s patogeny s MIC 4 mcg/ml, by měla být při hodnocení klinické odpovědi zvážena nižší systémová expozice, místo a závažnost infekce a základní zdravotní stav [viz KLINICKÁ FARMAKOLOGIE a DÁVKOVÁNÍ A PODÁVÁNÍ ].
Geriatrické použití
2 046 pacientů léčených přípravkem ZYVOX 600 mg ve fázi 3 klinických studií kontrolovaných komparátorem bylo 589 (29 %) starších 65 let a 253 (12 %) bylo 75 let nebo více. Mezi těmito pacienty a mladšími pacienty nebyly pozorovány žádné celkové rozdíly v bezpečnosti nebo účinnosti a další hlášené klinické zkušenosti neidentifikovaly rozdíly v odpovědích mezi staršími a mladšími pacienty, nelze však vyloučit větší citlivost některých starších jedinců.
PŘEDÁVKOVAT
případě předávkování se doporučuje podpůrná péče se zachováním glomerulární filtrace. Hemodialýza může usnadnit rychlejší eliminaci linezolidu. V klinické studii fáze 1 bylo přibližně 30 % dávky linezolidu odstraněno během 3hodinové hemodialýzy začínající 3 hodiny po podání dávky linezolidu. Údaje o odstranění linezolidu peritoneální dialýzou nebo hemoperfuzí nejsou k dispozici. Klinické příznaky akutní toxicity u zvířat byly snížená aktivita a ataxie u potkanů a zvracení a třes u psů léčených dávkou 3 000 mg/kg/den a 2 000 mg/kg/den.
KONTRAINDIKACE
Přecitlivělost
Formulace ZYVOX jsou kontraindikovány pro použití u pacientů, kteří mají známou přecitlivělost na linezolid nebo na kteroukoli další složku přípravku.
Inhibitory monoaminoxidázy
Linezolid by neměli užívat pacienti užívající jakýkoli léčivý přípravek, který inhibuje monoaminooxidázy A nebo B (např. fenelzin, isokarboxazid), nebo během dvou týdnů po užití takového léčivého přípravku.
KLINICKÁ FARMAKOLOGIE
Mechanismus působení
ZYVOX je antibakteriální léčivo [viz Mikrobiologie ].
Farmakodynamika
randomizované, pozitivně a placebem kontrolované zkřížené důkladné QT studii byla 40 zdravým subjektům podána jedna dávka ZYVOX 600 mg prostřednictvím 1hodinové IV infuze, jedna dávka ZYVOX 1 200 mg prostřednictvím 1hodinové IV infuze, placebo a jedna perorální dávka pozitivní kontroly. U obou dávek 600 mg a 1 200 mg ZYVOX 600 mg nebyl zjištěn žádný významný účinek na QTc interval při maximální plazmatické koncentraci ani v žádném jiném čase.
Farmakokinetika
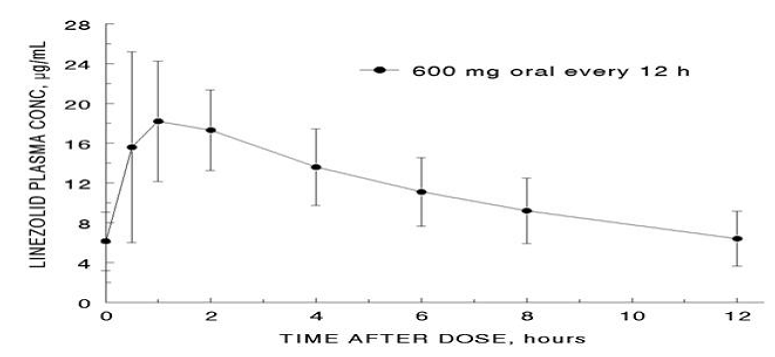
Průměrné farmakokinetické parametry linezolidu u dospělých po jednorázové a opakované perorální a intravenózní dávce jsou shrnuty v tabulce 8. Plazmatické koncentrace linezolidu v ustáleném stavu po perorálních dávkách 600 mg podávaných každých 12 hodin jsou uvedeny na obrázku 1.
Obrázek 1. Plazmatické koncentrace linezolidu u dospělých v ustáleném stavu po perorálním podání každých 12 hodin (průměr ± standardní odchylka, n=16)
Vstřebávání
Linezolid je po perorálním podání značně absorbován. Maximální plazmatické koncentrace je dosaženo přibližně za 1 až 2 hodiny po podání a absolutní biologická dostupnost je přibližně 100 %. Proto může být linezolid podáván perorálně nebo intravenózně bez úpravy dávky.
Linezolid lze podávat bez ohledu na načasování jídla. Doba k dosažení maximální koncentrace je zpožděna z 1,5 hodiny na 2,2 hodiny a Cmax se sníží asi o 17 %, když se s linezolidem podává jídlo s vysokým obsahem tuku. Celková expozice měřená jako AUC0-∞ je však za obou podmínek podobná.
Rozdělení
Farmakokinetické studie na zvířatech a lidech prokázaly, že linezolid se snadno distribuuje do dobře prokrvených tkání. Vazba linezolidu na plazmatické proteiny je přibližně 31 % a je nezávislá na koncentraci. Distribuční objem linezolidu v ustáleném stavu byl u zdravých dospělých dobrovolníků v průměru 40 až 50 litrů.
Koncentrace linezolidu byly stanoveny v různých tekutinách od omezeného počtu subjektů ve fázi 1 studií s dobrovolníky po opakovaném podávání linezolidu. Poměr linezolidu ve slinách k plazmě byl 1,2 ku 1 a poměr linezolidu v potu k plazmě byl 0,55 ku 1.
Metabolismus
Linezolid je primárně metabolizován oxidací morfolinového kruhu, což vede ke vzniku dvou neaktivních metabolitů karboxylových kyselin s otevřeným kruhem: metabolitu kyseliny aminoethoxyoctové (A) a metabolitu hydroxyethylglycinu (B). Předpokládá se, že tvorba metabolitu A vzniká enzymatickou cestou, zatímco metabolit B je zprostředkován neenzymatickým chemickým oxidačním mechanismem in vitro. Studie in vitro prokázaly, že linezolid je minimálně metabolizován a může být zprostředkován lidským cytochromem P450. Metabolická dráha linezolidu však není zcela objasněna.
Vylučování
Nerenální clearance představuje přibližně 65 % celkové clearance linezolidu. Za ustálených podmínek se přibližně 30 % dávky objeví v moči jako linezolid, 40 % jako metabolit B a 10 % jako metabolit A. Průměrná renální clearance linezolidu je 40 ml/min, což naznačuje čistou tubulární reabsorpci. Prakticky žádný linezolid se neobjevuje ve stolici, zatímco přibližně 6 % dávky se objevuje ve stolici jako metabolit B a 3 % jako metabolit A.
Se zvyšujícími se dávkami linezolidu byl pozorován malý stupeň nelinearity v clearance, což se zdá být způsobeno nižší renální a nerenální clearance linezolidu při vyšších koncentracích. Rozdíl v clearance byl však malý a neodrážel se ve zdánlivém poločasu eliminace.
Specifické populace
Geriatričtí pacienti
Farmakokinetika linezolidu se u starších pacientů (65 let nebo starších) významně nemění. Úprava dávky u geriatrických pacientů proto není nutná.
Pediatričtí pacienti
Farmakokinetika linezolidu po jednorázové intravenózní dávce byla zkoumána u pediatrických pacientů ve věku od narození do 17 let (včetně nedonošených a donošených novorozenců), u zdravých dospívajících jedinců ve věku od 12 do 17 let au pediatrických pacientů v rozmezí ve věku od 1 týdne do 12 let. Farmakokinetické parametry linezolidu jsou shrnuty v tabulce 9 pro studované pediatrické populace a zdravé dospělé subjekty po podání jedné intravenózní dávky.
Cmax a distribuční objem (Vss) linezolidu jsou u pediatrických pacientů podobné bez ohledu na věk. Plazmatická clearance linezolidu se však mění v závislosti na věku. S vyloučením předčasně narozených novorozenců mladších než jeden týden je clearance založená na hmotnosti nejrychlejší u nejmladších věkových skupin v rozmezí od
Podobné průměrné denní hodnoty AUC byly pozorovány u pediatrických pacientů od narození do 11 let věku, kterým byla podávána dávka každých 8 hodin, ve srovnání s dospívajícími nebo dospělými, kteří dostávali dávku každých 12 hodin. Proto by dávka pro pediatrické pacienty do 11 let měla být 10 mg/kg každých 8 hodin. Pediatričtí pacienti ve věku 12 let a starší by měli dostávat 600 mg každých 12 hodin [viz DÁVKOVÁNÍ A PODÁVÁNÍ ].
Rod
Ženy mají mírně nižší distribuční objem linezolidu než muži. Plazmatické koncentrace jsou vyšší u žen než u mužů, což je částečně způsobeno rozdíly v tělesné hmotnosti. Po dávce 600 mg je průměrná perorální clearance přibližně o 38 % nižší u žen než u mužů. Neexistují však žádné významné rozdíly mezi pohlavími v průměrné zdánlivé rychlostní konstantě nebo poločasu eliminace. Neočekává se tedy, že by se expozice léku u žen podstatně zvýšila nad úrovně, o nichž je známo, že jsou dobře tolerovány. Úprava dávky podle pohlaví se proto nezdá být nutná.
Renální poškození
Farmakokinetika původního léčiva, linezolidu, se u pacientů s jakýmkoliv stupněm poruchy funkce ledvin nemění; dva primární metabolity linezolidu se však akumulují u pacientů s poruchou funkce ledvin, přičemž množství akumulace se zvyšuje se závažností renální dysfunkce (viz tabulka 10). Farmakokinetika linezolidu a jeho dvou metabolitů byla také studována u pacientů s terminálním onemocněním ledvin (ESRD), kteří podstupují hemodialýzu. Ve studii ESRD byl 14 pacientům podáván linezolid v dávce 600 mg každých 12 hodin po dobu 14,5 dne (viz tabulka 11). Vzhledem k tomu, že podobných plazmatických koncentrací linezolidu je dosahováno bez ohledu na funkci ledvin, není u pacientů s poruchou funkce ledvin doporučena žádná úprava dávky. Vzhledem k absenci informací o klinickém významu akumulace primárních metabolitů by však použití linezolidu u pacientů s poruchou funkce ledvin mělo být zváženo oproti potenciálnímu riziku akumulace těchto metabolitů. Linezolid i oba metabolity jsou eliminovány hemodialýzou. Nejsou dostupné žádné informace o účinku peritoneální dialýzy na farmakokinetiku linezolidu. Přibližně 30 % dávky bylo eliminováno při 3hodinové hemodialýze začínající 3 hodiny po podání dávky linezolidu; proto by měl být linezolid podáván po hemodialýze.
Poškození jater
Farmakokinetika linezolidu se u pacientů (n=7) s mírnou až středně těžkou poruchou funkce jater (Child-Pugh třída A nebo B) nemění. Na základě dostupných informací se u pacientů s mírnou až středně těžkou poruchou funkce jater nedoporučuje žádná úprava dávkování. Farmakokinetika linezolidu u pacientů s těžkou poruchou funkce jater nebyla hodnocena.
Lékové interakce
Léky metabolizované cytochromem P450
Linezolid není induktorem cytochromu P450 (CYP450) u potkanů. Linezolid navíc neinhibuje aktivity klinicky významných lidských izoforem CYP (např. 1A2, 2C9, 2C19, 2D6, 2E1, 3A4). Proto se neočekává, že by linezolid ovlivňoval farmakokinetiku jiných léků metabolizovaných těmito hlavními enzymy. Současné podávání linezolidu podstatně nemění farmakokinetické charakteristiky (S)-warfarinu, který je rozsáhle metabolizován CYP2C9. Léky jako warfarin a fenytoin, které jsou substráty CYP2C9, mohou být podávány s linezolidem beze změn v dávkovacím režimu.
Antibakteriální léky
Aztreonam: Farmakokinetika linezolidu nebo aztreonamu se při společném podávání nemění.
Gentamicin: Farmakokinetika linezolidu nebo gentamicinu se při společném podávání nemění.
Antioxidanty
Potenciál lékových interakcí s linezolidem a antioxidanty vitaminem C a vitaminem E byl studován u zdravých dobrovolníků. Subjektům byla podávána perorální dávka 600 mg linezolidu v den 1 a další dávka 600 mg linezolidu v den 8. Ve dnech 2-9 byl subjektům podáván buď vitamin C (1 000 mg/den) nebo vitamin E (800 IU/den). den). AUC0-∞ linezolidu se zvýšila o 2,3 % při současném podávání s vitaminem C a o 10,9 % při současném podávání s vitaminem E. Při současném podávání s vitaminem C nebo vitaminem E se nedoporučuje žádná úprava dávky linezolidu.
Silné induktory CYP 3A4
rifampin
Účinek rifampinu na farmakokinetiku linezolidu byl hodnocen ve studii 16 zdravých dospělých mužů. Dobrovolníkům byl perorálně podáván linezolid 600 mg dvakrát denně v 5 dávkách s rifampinem a bez rifampinu 600 mg jednou denně po dobu 8 dnů. Současné podávání rifampinu s linezolidem vedlo k 21% snížení Cmax linezolidu [90% CI, 15% - 27%] a 32% snížení AUC0-12 linezolidu [90% CI, 27% - 37%]. Klinický význam této interakce není znám. Mechanismus této interakce není plně objasněn a může souviset s indukcí jaterních enzymů. Jiné silné induktory jaterních enzymů (např. karbamazepin, fenytoin, fenobarbital) by mohly způsobit podobné nebo menší snížení expozice linezolidu.
Inhibice monoaminoxidázy
Linezolid je reverzibilní, neselektivní inhibitor monoaminooxidázy. Proto má linezolid potenciál pro interakci s adrenergními a serotonergními látkami.
Adrenergní látky
některých jedinců užívajících ZYVOX 600 mg může dojít k reverzibilnímu zesílení presorické odpovědi na nepřímo působící sympatomimetika, vazopresorická nebo dopaminergní látky. Specificky byly studovány běžně používané léky, jako je fenylpropanolamin a pseudoefedrin. Počáteční dávky adrenergních látek, jako je dopamin nebo epinefrin, by měly být sníženy a titrovány, aby se dosáhlo požadované odpovědi.
tyramin
Významná presorická odpověď byla pozorována u normálních dospělých subjektů, kterým byly podávány dávky linezolidu a tyraminu vyšší než 100 mg. Pacienti užívající linezolid se proto musí vyvarovat konzumace velkého množství potravin nebo nápojů s vysokým obsahem tyraminu [viz INFORMACE PRO PACIENTA ].
Pseudoefedrin HCl nebo fenylpropanolamin HCl
Když je linezolid podáván zdravým normotenzním subjektům, je pozorováno reverzibilní zvýšení presorické odpovědi buď pseudoefedrinu HCl (PSE) nebo fenylpropanolaminu HCl (PPA) [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ a DROGOVÉ INTERAKCE ]. Podobná studie nebyla provedena u pacientů s hypertenzí. Interakční studie provedené u normotenzních subjektů hodnotily účinky na krevní tlak a srdeční frekvenci placeba, samotného PPA nebo PSE, samotného linezolidu a kombinace linezolidu v ustáleném stavu (600 mg každých 12 hodin po dobu 3 dnů) se dvěma dávkami PPA ( 25 mg) nebo PSE (60 mg) podaných s odstupem 4 hodin. Srdeční frekvence nebyla ovlivněna žádnou léčbou. Krevní tlak byl zvýšen při obou kombinovaných léčbách. Maximální hladiny krevního tlaku byly pozorovány 2 až 3 hodiny po druhé dávce PPA nebo PSE a vrátily se na výchozí hodnotu 2 až 3 hodiny po vrcholu. Následují výsledky studie PPA ukazující průměrný (a rozsah) maximální systolický krevní tlak v mm Hg: placebo = 121 (103 až 158); samotný linezolid = 120 (107 až 135); samotná PPA = 125 (106 až 139); PPA s linezolidem = 147 (129 až 176). Výsledky studie PSE byly podobné jako výsledky studie PPA. Průměrné maximální zvýšení systolického krevního tlaku nad výchozí hodnotu bylo 32 mm Hg (rozmezí: 20-52 mm Hg) a 38 mm Hg (rozsah: 18-79 mm Hg) během současného podávání linezolidu s pseudoefedrinem nebo fenylpropanolaminem, v daném pořadí.
Serotonergní látky
Dextromethorfan
Potenciální léková interakce s dextromethorfanem byla studována u zdravých dobrovolníků. Subjektům byl podáván dextromethorfan (dvě 20mg dávky podané s odstupem 4 hodin) s linezolidem nebo bez něj. U normálních jedinců užívajících linezolid a dextromethorfan nebyly pozorovány žádné účinky serotoninového syndromu (zmatenost, delirium, neklid, třes, ruměnec, pocení, hyperpyrexie).
Mikrobiologie
Mechanismus působení
Linezolid je syntetické antibakteriální činidlo ze třídy oxazolidinonů, které má klinickou využitelnost při léčbě infekcí způsobených aerobními grampozitivními bakteriemi. Spektrum in vitro aktivity linezolidu také zahrnuje určité gramnegativní bakterie a anaerobní bakterie. Linezolid se váže na místo na bakteriální 23S ribozomální RNA podjednotky 50S a zabraňuje tvorbě funkčního iniciačního komplexu 70S, který je nezbytný pro reprodukci bakterií. Výsledky časových studií ukázaly, že linezolid je bakteriostatický proti enterokokům a stafylokokům. U streptokoků bylo zjištěno, že linezolid je baktericidní pro většinu izolátů.
Odpor
Studie in vitro ukázaly, že bodové mutace v 23S rRNA jsou spojeny s rezistencí na linezolid. Byly publikovány zprávy o tom, že se Enterococcus faecium rezistentní na vankomycin stal rezistentním vůči linezolidu během jeho klinického použití. Existují zprávy o tom, že Staphylococcus aureus (rezistentní na meticilin) během klinického používání vyvine rezistenci na linezolid. Rezistence na linezolid u těchto organismů je spojena s bodovou mutací v 23S rRNA (substituce thyminu za guanin na pozici 2576) organismu. Organismy rezistentní na oxazolidinony prostřednictvím mutací v chromozomálních genech kódujících 23S rRNA nebo ribozomální proteiny (L3 a L4) jsou obecně zkříženě rezistentní na linezolid. Rovněž byla hlášena rezistence na linezolid u stafylokoků zprostředkovaná enzymem methyltransferázou. Tato rezistence je zprostředkována genem cfr (chloramfenikol-florfenikol) umístěným na plazmidu, který je přenosný mezi stafylokoky.
Interakce s jinými antimikrobiálními léky
Studie in vitro prokázaly aditivitu nebo indiferenci mezi linezolidem a vankomycinem, gentamicinem, rifampinem, imipenem-cilastatinem, aztreonamem, ampicilinem nebo streptomycinem.
Bylo prokázáno, že linezolid je účinný proti většině izolátů následujících mikroorganismů, a to jak in vitro, tak při klinických infekcích (viz INDIKACE ].
Gram-pozitivní bakterie
Enterococcus faecium (pouze izoláty rezistentní na vankomycin) Staphylococcus aureus (včetně izolátů rezistentních na meticilin) Streptococcus agalactiae Streptococcus pneumoniae Streptococcus pyogenes
K dispozici jsou následující údaje in vitro, ale jejich klinický význam není znám. Více než 90 % následujících bakterií vykazuje in vitro MIC menší nebo rovnou hraničnímu bodu citlivosti na linezolid pro organismy podobného rodu. Bezpečnost a účinnost linezolidu při léčbě klinických infekcí způsobených těmito bakteriemi nebyla stanovena v adekvátních a dobře kontrolovaných klinických studiích.
Gram-pozitivní bakterie
Enterococcus faecalis (včetně izolátů rezistentních na vankomycin) Enterococcus faecium (izoláty citlivé na vankomycin) Staphylococcus epidermidis (včetně izolátů rezistentních na meticilin) Streptokoky skupiny Staphylococcus haemolyticus Viridans
Gram-negativní bakterie
Pasteurella multocida
Testování citlivosti
Konkrétní informace týkající se interpretačních kritérií testu citlivosti a souvisejících testovacích metod a standardů kontroly kvality uznaných FDA pro tento lék naleznete na: https://www.fda.gov/STIC.
Toxikologie zvířat a/nebo farmakologie
Cílové orgány toxicity linezolidu byly podobné u juvenilních a dospělých potkanů a psů. Ve studiích na zvířatech byla pozorována myelosuprese závislá na dávce a čase, o čemž svědčí hypocelularita kostní dřeně/snížená hematopoéza, snížená extramedulární hematopoéza ve slezině a játrech a snížené hladiny cirkulujících erytrocytů, leukocytů a krevních destiček. Lymfoidní deplece se objevila v brzlíku, lymfatických uzlinách a slezině. Obecně byly lymfoidní nálezy spojeny s anorexií, ztrátou hmotnosti a potlačením nárůstu tělesné hmotnosti, což mohlo přispět k pozorovaným účinkům.
potkanů, kterým byl linezolid podáván perorálně po dobu 6 měsíců, byla pozorována nereverzibilní, minimální až mírná axonální degenerace sedacích nervů při dávce 80 mg/kg/den; minimální degenerace sedacího nervu byla také pozorována u 1 muže při této hladině dávky při 3měsíční prozatímní nekropsii. Bylo provedeno citlivé morfologické hodnocení perfuzně fixovaných tkání, aby se prozkoumaly známky degenerace zrakového nervu. Minimální až střední degenerace zrakového nervu byla evidentní u 2 samců potkanů po 6 měsících podávání, ale přímý vztah k léku byl nejednoznačný kvůli akutní povaze nálezu a jeho asymetrické distribuci. Pozorovaná degenerace nervu byla mikroskopicky srovnatelná se spontánní jednostrannou degenerací zrakového nervu hlášenou u stárnoucích potkanů a může být exacerbací běžné změny pozadí.
Tyto účinky byly pozorovány při hladinách expozice, které jsou srovnatelné s hladinami pozorovanými u některých lidských subjektů. Hematopoetické a lymfoidní účinky byly reverzibilní, i když v některých studiích byla reverze neúplná během období zotavení.
Klinické studie
Dospělí
Nozokomiální pneumonie
Do randomizované, multicentrické, dvojitě zaslepené studie byli zařazeni dospělí pacienti s klinicky a radiologicky dokumentovanou nozokomiální pneumonií. Pacienti byli léčeni po dobu 7 až 21 dnů. Jedna skupina dostávala ZYVOX IV injekci 600 mg každých 12 hodin a druhá skupina dostávala vankomycin 1 g každých 12 hodin intravenózně. Obě skupiny dostávaly souběžně aztreonam (1 až 2 g každých 8 hodin intravenózně), v němž bylo možné pokračovat, pokud je to klinicky indikováno. Do studie bylo zařazeno 203 pacientů léčených linezolidem a 193 pacientů léčených vankomycinem. Klinicky hodnotitelných bylo sto dvacet dva (60 %) pacientů léčených linezolidem a 103 (53 %) pacientů léčených vankomycinem. Míra vyléčení u klinicky hodnotitelných pacientů byla 57 % u pacientů léčených linezolidem a 60 % u pacientů léčených vankomycinem. Míra vyléčení u klinicky hodnotitelných pacientů s ventilátorovou pneumonií byla 47 % u pacientů léčených linezolidem a 40 % u pacientů léčených vankomycinem. Modifikovaný intent-to-treat (MITT) analýza 94 pacientů léčených linezolidem a 83 pacientů léčených vankomycinem zahrnovala subjekty, u kterých byl před léčbou izolován patogen. Míra vyléčení v analýze MITT byla 57 % u pacientů léčených linezolidem a 46 % u pacientů léčených vankomycinem. Míry vyléčení podle patogenu u mikrobiologicky hodnotitelných pacientů jsou uvedeny v tabulce 12.
Komplikované infekce kůže a kožní struktury
Dospělí pacienti s klinicky zdokumentovanými komplikovanými infekcemi kůže a kožní struktury byli zařazeni do randomizované, multicentrické, dvojitě zaslepené, dvojitě slepé studie srovnávající studované léky podávané intravenózně a následně léky podávané perorálně po dobu celkem 10 až 21 dnů léčby. Jedna skupina pacientů dostávala ZYVOX IV injekci 600 mg každých 12 hodin a následně ZYVOX 600 mg tablety 600 mg každých 12 hodin; druhá skupina dostávala oxacilin 2 g každých 6 hodin intravenózně a následně dikloxacilin 500 mg každých 6 hodin perorálně. Pacienti by mohli současně dostávat aztreonam, pokud je to klinicky indikováno. Do studie bylo zařazeno 400 pacientů léčených linezolidem a 419 pacientů léčených oxacilinem. Dvě stě čtyřicet pět (61 %) pacientů léčených linezolidem a 242 (58 %) pacientů léčených oxacilinem bylo klinicky hodnotitelných. Míra vyléčení u klinicky hodnotitelných pacientů byla 90 % u pacientů léčených linezolidem a 85 % u pacientů léčených oxacilinem. Modifikovaná analýza záměru léčby (MITT) 316 pacientů léčených linezolidem a 313 pacientů léčených oxacilinem zahrnovala subjekty, které splnily všechna kritéria pro zařazení do studie. Míra vyléčení v analýze MITT byla 86 % u pacientů léčených linezolidem a 82 % u pacientů léčených oxacilinem. Míry vyléčení podle patogenu u mikrobiologicky hodnotitelných pacientů jsou uvedeny v tabulce 13.
Další zkušenosti s použitím přípravku ZYVOX v léčbě infekcí Staphylococcus aureus (MRSA) rezistentních na meticilin poskytla samostatná studie. Jednalo se o randomizovanou, otevřenou studii u hospitalizovaných dospělých pacientů s dokumentovanou nebo suspektní infekcí MRSA.
Jedna skupina pacientů dostávala ZYVOX IV injekci 600 mg každých 12 hodin a následně tablety ZYVOX 600 mg každých 12 hodin. Druhá skupina pacientů dostávala vankomycin 1 g každých 12 hodin intravenózně. Obě skupiny byly léčeny po dobu 7 až 28 dnů a mohly současně dostávat aztreonam nebo gentamicin, pokud je to klinicky indikováno. Míra vyléčení u mikrobiologicky hodnotitelných pacientů s infekcí kůže a kožních struktur MRSA byla 26/33 (79 %) u pacientů léčených linezolidem a 24/33 (73 %) u pacientů léčených vankomycinem.
Infekce diabetické nohy
Dospělí diabetici s klinicky zdokumentovanými komplikovanými infekcemi kůže a kožní struktury („diabetické infekce nohou“) byli zařazeni do randomizované (poměr 2:1), multicentrické, otevřené studie porovnávající studované léky podávané intravenózně nebo perorálně v celkovém počtu 14 až 28 dnů léčby. Jedna skupina pacientů dostávala ZYVOX 600 mg každých 12 hodin intravenózně nebo perorálně; druhá skupina dostávala ampicilin/sulbaktam 1,5 až 3 g intravenózně nebo amoxicilin/klavulanát 500 až 875 mg každých 8 až 12 hodin perorálně. V zemích, kde ampicilin/sulbaktam není na trhu, byl pro intravenózní režim použit amoxicilin/klavulanát v dávce 500 mg až 2 g každých 6 hodin. Pacienti ve srovnávací skupině mohli být také léčeni vankomycinem 1 g každých 12 hodin intravenózně, pokud byl MRSA izolován z infekce nohy. Pacienti v obou léčebných skupinách, kteří měli gramnegativní bacily izolované z místa infekce, mohli také dostávat aztreonam 1 až 2 g každých 8-12 hodin intravenózně. Všichni pacienti měli nárok na vhodné doplňkové léčebné metody, jako je debridement a vyložení, jak je obvykle vyžadováno při léčbě infekcí diabetické nohy, a většina pacientů tuto léčbu dostávala. V populaci studie intent-to-treat (ITT) bylo 241 pacientů léčených linezolidem a 120 pacientů léčených komparátorem. Dvě stě dvanáct (86 %) pacientů léčených linezolidem a 105 (85 %) pacientů léčených komparátorem bylo klinicky hodnotitelných. V populaci ITT byla míra vyléčení 68,5 % (165/241) u pacientů léčených linezolidem a 64 % (77/120) u pacientů léčených komparátorem, kde byli pacienti s neurčitými a chybějícími výsledky považováni za neúspěšné. Míra vyléčení u klinicky hodnotitelných pacientů (kromě pacientů s neurčitými a chybějícími výsledky) byla 83 % (159/192) a 73 % (74/101) u pacientů léčených linezolidem a komparátorem. Kritická post-hoc analýza se zaměřila na 121 pacientů léčených linezolidem a 60 pacientů léčených komparátorem, kteří měli grampozitivní patogen izolovaný z místa infekce nebo z krve, kteří měli méně známek základní osteomyelitidy než celková studovaná populace a kteří nedostali zakázané antimikrobiální látky. Na základě této analýzy byla míra vyléčení 71 % (86/121) u pacientů léčených linezolidem a 63 % (38/60) u pacientů léčených komparátorem. Žádná z výše uvedených analýz nebyla upravena pro použití doplňkových terapií. Míry vyléčení podle patogenu u mikrobiologicky hodnotitelných pacientů jsou uvedeny v tabulce 14.
Enterokokové infekce rezistentní na vankomycin
Dospělí pacienti s dokumentovanou nebo suspektní enterokokovou infekcí rezistentní na vankomycin byli zařazeni do randomizované, multicentrické, dvojitě zaslepené studie porovnávající vysokou dávku ZYVOXu (600 mg) s nízkou dávkou ZYVOXu (200 mg) podávanou každých 12 hodin. intravenózně (IV) nebo perorálně po dobu 7 až 28 dnů. Pacienti mohli současně dostávat aztreonam nebo aminoglykosidy. Bylo randomizováno 79 pacientů k vysoké dávce linezolidu a 66 k nízké dávce linezolidu. Populace intent-to-treat (ITT) s dokumentovanou enterokokovou infekcí rezistentní na vankomycin na začátku sestávala z 65 pacientů ve větvi s vysokou dávkou a 52 ve větvi s nízkou dávkou.
Míry vyléčení pro populaci ITT s dokumentovanou enterokokovou infekcí rezistentní na vankomycin na začátku studie jsou uvedeny v tabulce 15 podle zdroje infekce. Tato míra vyléčení nezahrnuje pacienty s chybějícími nebo neurčitými výsledky. Míra vyléčení byla vyšší ve větvi s vysokou dávkou než ve větvi s nízkou dávkou, ačkoli rozdíl nebyl statisticky významný na úrovni 0,05.
Pediatričtí pacienti
Infekce způsobené grampozitivními bakteriemi
Studie bezpečnosti a účinnosti poskytla zkušenosti s použitím přípravku ZYVOX 600 mg u dětských pacientů k léčbě nozokomiální pneumonie, komplikovaných infekcí kůže a kožních struktur a dalších infekcí způsobených grampozitivními bakteriálními patogeny, včetně methicilin-rezistentního a citlivého Staphylococcus aureus a Enterococcus faecium rezistentní na vankomycin. Pediatričtí pacienti ve věku od narození do 11 let s infekcemi způsobenými zdokumentovanými nebo suspektními grampozitivními bakteriemi byli zařazeni do randomizované studie s označením openPage kontrolované komparátorem. Jedna skupina pacientů dostávala ZYVOX 600 mg IV injekce 10 mg/kg každých 8 hodin a následně ZYVOX 600 mg pro perorální suspenzi 10 mg/kg každých 8 hodin. Druhá skupina dostávala vankomycin v dávce 10 až 15 mg/kg intravenózně každých 6 až 24 hodin v závislosti na věku a renální clearance. Pacienti, u kterých byla potvrzena infekce VRE, byli zařazeni do třetí větve studie a dostávali ZYVOX 10 mg/kg každých 8 hodin intravenózně a/nebo perorálně. Všichni pacienti byli léčeni celkem 10 až 28 dní a mohli současně dostávat gramnegativní antibakteriální léky, pokud je to klinicky indikováno. V populaci intent-to-treat (ITT) bylo 206 pacientů randomizovaných na linezolid a 102 pacientů randomizovaných na vankomycin. Míry vyléčení u pacientů s ITT, MITT a klinicky hodnotitelných pacientů jsou uvedeny v tabulce 16. Po dokončení studie bylo 13 dalších pacientů ve věku od 4 dnů do 16 let zařazeno do otevřeného rozšíření ramene VRE studie. Tabulka 17 uvádí míru klinického vyléčení podle patogenu pro mikrobiologicky hodnotitelné pacienty včetně mikrobiologicky hodnotitelných pacientů s Enterococcus faecium rezistentním na vankomycin z rozšíření této studie.
INFORMACE PRO PACIENTA
Důležité pokyny pro administraci
Informujte pacienty, že ZYVOX lze užívat s jídlem nebo bez jídla.
Periferní a optická neuropatie
Informujte pacienty, aby informovali svého lékaře, pokud během užívání ZYVOXu zaznamenají změny vidění [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
serotoninový syndrom
Informujte pacienty, aby informovali svého lékaře, pokud užívají serotonergní látky, včetně inhibitorů zpětného vychytávání serotoninu nebo jiných antidepresiv a opioidů (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Potenciální interakce způsobující zvýšení krevního tlaku
- Doporučte pacientům, aby informovali svého lékaře, pokud mají v anamnéze hypertenzi.
- Doporučte pacientům, aby se během užívání přípravku ZYVOX vyhýbali velkému množství potravin nebo nápojů s vysokým obsahem tyraminu. Potraviny s vysokým obsahem tyraminu zahrnují ty, které mohly projít změnami bílkovin stárnutím, fermentací, nakládáním nebo uzením za účelem zlepšení chuti, jako jsou stařené sýry, fermentované nebo sušené maso, kysané zelí, sójová omáčka, čepovaná piva a červená vína. Obsah tyraminu v jakékoli potravině bohaté na bílkoviny se může zvýšit, pokud je skladována po dlouhou dobu nebo je nesprávně chlazena.
- Poraďte pacientům, aby informovali svého lékaře, pokud užívají léky obsahující pseudoefedrin HCl nebo fenylpropanolamin HCl, jako jsou léky na nachlazení a dekongestanty [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Laktátová acidóza
Informujte pacienty, aby informovali svého lékaře, pokud se u nich během užívání ZYVOXu objeví opakované epizody nevolnosti nebo zvracení [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Křeče
Poraďte pacientům, aby informovali svého lékaře, pokud mají v anamnéze záchvaty nebo křeče [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Hypoglykémie
Doporučte pacientům, aby informovali svého lékaře, pokud mají diabetes mellitus. Při léčbě linezolidem se mohou objevit hypoglykemické reakce, jako je pocení a třes, spolu s nízkou hladinou glukózy v krvi. Pokud se takové reakce objeví, pacienti by měli kontaktovat lékaře nebo jiného zdravotnického pracovníka pro správnou léčbu [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Hyponatremie a/nebo SIADH
Poraďte pacientům s rizikem hyponatremie, aby informovali svého lékaře, pokud se u nich objeví známky a příznaky hyponatremie a/nebo SIADH, včetně zmatenosti, ospalosti, celkové slabosti a respirační tísně (viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Fenylketonurie
Informujte pacienty s fenylketonurií (PKU), že každých 5 ml přípravku ZYVOX pro perorální suspenzi 100 mg/5 ml obsahuje 20 mg fenylalaninu. Ostatní přípravky ZYVOX 600 mg fenylalanin neobsahují. Fenylalanin může být škodlivý pro pacienty s fenylketonurií. Kontaktujte svého lékaře nebo lékárníka, pokud je předepsán přípravek ZYVOX 600 mg pro perorální suspenzi [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Antibakteriální odolnost
Pacienti by měli být poučeni, že antibakteriální léky včetně ZYVOXu by se měly používat pouze k léčbě bakteriálních infekcí. Neléčí virové infekce (např. nachlazení). Pokud je ZYVOX 600 mg předepisován k léčbě bakteriální infekce, je třeba pacientům říci, že ačkoli je běžné, že se na začátku léčby cítí lépe, lék by měl být užíván přesně podle pokynů. Vynechání dávek nebo nedokončení celé terapie může snížit účinnost okamžité léčby a zvýšit pravděpodobnost, že si bakterie vyvinou rezistenci a nebudou v budoucnu léčitelné ZYVOXem nebo jinými antibakteriálními léky.
Průjem
Průjem je častým problémem způsobeným antibakteriálními léky, který obvykle končí vysazením antibakteriálního léku. Někdy po zahájení léčby antibakteriálními léky se u pacientů může objevit vodnatá a krvavá stolice (s žaludečními křečemi a horečkou nebo bez nich) dokonce až dva nebo více měsíců po užití poslední dávky antibakteriálního léku. Pokud k tomu dojde, pacienti by měli co nejdříve kontaktovat svého lékaře [viz VAROVÁNÍ A BEZPEČNOSTNÍ OPATŘENÍ ].
Neplodnost
Informujte pacienty mužského pohlaví, že ZYVOX může reverzibilně narušit plodnost [viz Použití u konkrétních populací ].
Označení tohoto produktu mohlo být aktualizováno. Nejnovější informace o předepisování naleznete na www.pfizer.com.